Development News Brief
Here are the highlights of the following upgrade:
hg pull -u -r 8729d2e29b02
What's New
Galaxy's FTP Server New Data Upload Option
- User how-to: UploadViaFTP
- Configuration instructions for local installs: wiki/Config/UploadViaFTP
OpenID Login
- User how-to and config instructions: wiki/OpenIDAuthentication
NGS Simulation Tool
-
Allows user to simulate multiple Illumina runs with several parameters that can be set.
- On each run, one position is randomly chosen to be polymorphic and sequencing errors are also simulated.
- The primary output is a png with two different plots.
- The other output shows summary statistics about the simulation.
- NGS simulation tool location: tools/ngs_simulation/ngs_simulation.xml
Tophat and Cufflinks RNA-seq Tools
-
Addition of RNA-seq analysis tools Tophat and Cufflinks.
- Together, these tools can be used to analyze RNA-seq data to understand alternative splicing and isoforms, gene and isoform expression, and perform statistical tests for differential expression.
- Galaxy supports Tophat version 1.1.1 and later and Cufflinks version 0.9.1 and later. (These are the versions included this distribution).
Import or Export Workflows & Histories
- Workflows can now be downloaded/exported to a file and uploaded/imported into Galaxy, making it easy to move workflows between Galaxy instances.
-
Beta feature: Histories can also be downloaded or moved from one Galaxy instance to another, subject to these limitations:
- history archives can be uploaded/imported only via URL, not file
- histories must be accessible via link in order for them to be importable via archive
- tags are not currently imported
- reproducibility is limited as parameters for imported jobs are not always recovered and set
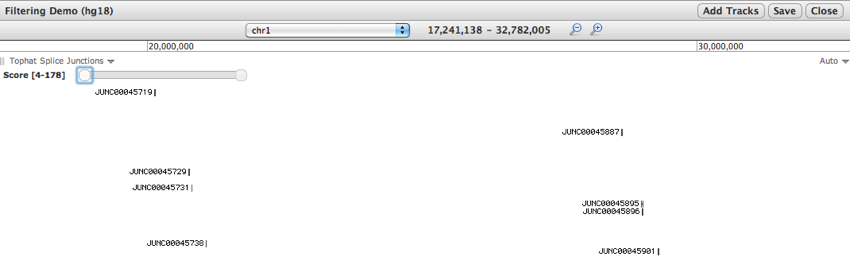
Even Better Data Visualization with Trackster
- Trackster now supports interactive filtering for VCF quality values and BED score values.
- For example, a user can drag a slider to filter a file of splice junctions to view junctions supported by different numbers of reads.
- Improved CIGAR support to BAM display. Properly displays matches,
deletions, skipped bases, and clipping. Padding for insertions are currently not represented in the display.
- GFF feature blocks are now displayed correctly, along with name, strand, and score information.
-
General enhancements
- Removed right-hand pane, allow inline re-ordering and configuration of elements
- Moved navigational controls to the top
- Histogram display for LineTracks and overview
- New navigational slider and new overview settings under the dropdown corresponding to the track name
- Summary view now shows maximum y-axis value
- Can change draw color of LineTrack
- When editing track config, "Enter" and "Esc" keys submit and cancel the changes, respectively
- Don't index bottom level for summary_tree, greatly reducing computation time (>5x speedup) while not sacrificing usability Refactored to pass JSLint
-
Tuning
- Fix ReferenceTrack issue.
- Don't re-add new datasets when refreshing after using "Add into current viz" link.
- To prevent browser lockup, only display up to 50 lines of features by default (user-editable in future). Coming soon: add warning message when this occurs.
- Fix LineTrack rendering bug when more than one tile on screen.
Native Data set Re-organization
- Galaxy now uses a set of data tables instead of simple loc files to organize, document, and store native genome data sets.
-
Why Data tables? Better data management for long term stability!
- Allows the information in the loc file, including the path, to be changed.
- By using a unique ID as the parameter value, data links in existing workflows are preserved.
- Most tools (PerM, Bowtie, BWA, Lastz, Megablast, SRMA, Tophat) that previously used loc files now have the new data tables organization implemented.
- Better data tracking has allowed for more informative genome name display in tool dropdown boxes.
-
For local installations:
- See the new wiki describing how to use data tables: [wiki/DataTables](/admin/tools/Data Tables/)
- More help for NGS tool setup (update pending): [wiki/NGSLocalSetup](/admin/NGS Local Setup/)
Updated & Improved
Sample Tracking
- Complete re-write of the Framework and User interface (database schema unchanged).
- New interactive interface to select files to transfer from the sequencer to Galaxy data libraries.
- The data transfer feature now uses Galaxy RESTful API.
- Full documentation detailing the new functionality and how to use it will be available within a few weeks through the home Galaxy Wiki.
Instantiating Galaxy
- New checkouts will now perform all necessary setup directly in run.sh, there is no longer a need to run setup.sh prior to run.sh. (setup.sh will be removed in a future distribution).
Analysis Tools
- Enable 'FASTX-Toolkit for FASTQ data' as a subsection under 'NGS: QC and manipulation' in tool_conf.xml.sample/main. Includes special handling for when the shell only allows for strict Bourne syntax.
- Add descriptive labels to output dataset names for MACS peakcalling tool.
- Taxonomy tools updated for better error reporting. Includes special handling for when the shell only allows for strict Bourne syntax.
- Refactor sam_bitwise_flag_filter tool, simplifying it and making it faster when there are multiple flag criteria
Tool Dependency Enhancements
- Addition of the 'package' type to
tags in the tool config. 1 Syntax for tool configs is:
<requirements>
<requirement type='package' version='X.Y.Z'>NAME</requirement>
</requirements>
2 Next, a directory should be created, and the path to that directory should be set in universe_wsgi.ini as 'tool_dependency_dir'.
3 Galaxy will then source the following file prior to executing the tool's
<tool_dependency_dir>/<NAME>/<X.Y.Z>/env.sh
4 The 'version' attribute of the 'requirement' tag is optional and if left off, Galaxy will look for the following instead:
<tool_dependency_dir>/<NAME>/default/env.sh
Data Libraries
- UI: new style for dropdown menus.
- Now uses jStore to save folder expansion state.
- Pre-generate and cache variables so that expensive functions like jQuery.siblings, jQuery.filter and jQuery.find only have to be called a minimum amount of times. Provides significant speedup to loading of large data libraries.
Genome Indexes
- Add basic support for Bowtie indexes as a datatype (bowtie_base_index, bowtie_color_index), available via datatype conversion. Currently, the indexes need to be converted manually from the FASTA file before use in Bowtie, but they can be reused.
- A new sample loc file (tool-data/all_fasta.loc.sample) was added which lists fasta files. A script (scripts/loc_files/create_all_fasta_loc.py) was created that can be used to generate this loc file for local installations.
Data Formats
- New gff2bed tool to convert GFF3 files to BED.
- Modified Filter and Sort -> Filter tool to operate correctly on files with a variable number of columns, such as in SAM files.
- New datatype added: VCF (variant call format).
Histories
- Add descriptive labels to output dataset names for MACS peakcalling tool.
- Add name/designation to HDA name for new datasets created in collect_primary_datasets.
Workflow Tuning
- Shift management of the interaction between workflow outputs and HideDatasetActions to the front end editor.
- No usability changes, but this resolves the issue with multiple HideDatasetActions being created.
- Existing workflows displaying multiple HideDatasetActions per step on the Run Workflow screen will persist. These extra HideDatasetActions are harmless, but a simple edit workflow -> save will remove them.
-
Workflow Inputs change:
- Workflow inputs that aren't a subtype of text, were previously not an option.
- Added 'data' datatype to registry, which will allow both text and binary inputs (and their subtypes) to workflow input steps.
- Note that this will allow a user to change the datatype of something to 'data'.
User Interface (UI)
- New function for downloading metadata files associated with datasets
(such as bai indices for bam files). See the Save icon drop-down menu.
- Enable display of unicode characters in history and workflow annotations and when listing and running workflows.
- Dynamicically generated popup-style menus. Greatly improves load
time, especially for data libraries having potentially large menu.
- Labels next to checkboxes can now be clicked to check the corresponding box.
- Radio boxes in tool forms now also have clickable labels as well.
- New style for search boxes in grids. Grid items will no longer show outline when hovered upon if there are no actions to be performed.
- Refactored refresh_on_change javascript code to run in galaxy.base when the page is loaded.
- Remove the creation of a background element that closes the active menu clicked. Instead, bind an event to close active menus to the document object of current and all other framesets. Tested in IE.
- Make links in split menu buttons "go through" instead of popping up the menu options.
General
- Functional Test Framework: new nose plugin that shows a diff between tests failed this time and last time.
- Documentation update to add more options added to the sample config file.
Bug Fixes!
- Fix for TextToolParameter.get_html_field when provided value is an empty string but default value specified in tool is non-empty string. Fixes issue with rerun button where if a user had input an empty string, the form displayed when rerun would have the default value from the tool and not the actual previously specified value.
- Fix for Integer/FloatToolParameter.get_html_field() when 'value' is provided as an integer/float. Fixes an issue seen when saving workflows: If an integer or float tool parameter is changed to a value of 0 or 0.0 and saved, the form field would be redisplayed using the default tool value; and not the value that is now saved in the database.
- Fix for setting columns in workflow builder for ColumnListParameter. e.g. allows splitting lists of columns by newlines and commas and strips leading 'c's.
- Fixes for rerun action to recurse grouping options when checking unvalidated values and cloned HDAs. Better selection of corresponding HDAs from cloned histories, when multiple copies exist.
- Have rerun action make use of tool.check_and_update_param_values(). Fixes Server Error issue when trying to rerun updated tools.
- Fix for display framework to work with workflows that contain tools that have been updated. Previously, this would cause a server error when trying to view a workflow or a page with an embedded workflow that contained an updated tool.
- Fix bug that was causing Page item selection grids to be initialized twice and hence causing grid paging to fail.
- Add some space between adjacent embedded items on Pages.
- Fix path to closebox.png image so screencast close button is shown correctly.
- Fix the Admin -> Manage Jobs interface when using multiple Galaxy processes
- When possible (e.g. Python >= 2.6), don't use tons of memory to handle zipped uploads.
- Fix cluster stdout/stderr handling that could cause excessive memory usage if stdout/stderr were very large.
- Make the PBS runner actually stop jobs when a user deletes output. This would only work before if the Galaxy user was a PBS "operator" and only using a single process setup.
- Cause waiting jobs to fail if any of their inputs fail to set metadata correctly.
- Fix 'import from current history' for Data Libraries that was showing metadata files that are not visible. Fix this same issue for 'Copy history items' feature.
- DRMAA runner now uses get_id_tag() in Wrapper instead of job_id directly for creation of .sh .o and .e files, as well as some debugging.
- Prevent Rename Dataset Action from allowing a blank input.
How to get Galaxy
hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist
About Galaxy
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State.