Development News Brief
- new:
% hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist - upgrade:
% hg pull -u -r 95d65755ac69
What's New
Workflow Additions
- Usability improvements for workflow annotations
- Workflow annotation is now shown at the top of the page.
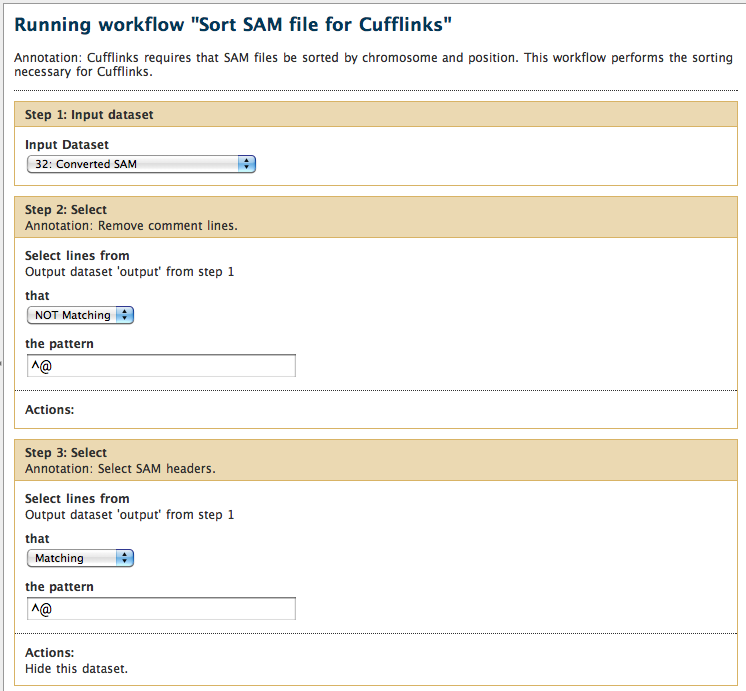
-
Step annotations are shown in the step header rather than at the bottom. example: Annotation
' width='746' height='691' xlink:href='data:image/png%3bbase64%2ciVBORw0KGgoAAAANSUhEUgAAAEAAAAA7CAIAAABE7huLAAAACXBIWXMAAAsTAAALEwEAmpwYAAAKt0lEQVRogdVa93PbRhbWn34/5G4yc8WWS5yJLx5f4sSSbKtZxZJMiRR7B0E0AiCJSqIQIED0fgfSVhRPbCt2YvK%2b%2bQbzZrkL4mF38d73gDVZlqMwjMIgisIkiYPAT%2bIwDAPP85IkiuNo/qsfBn6SRFEUWpbl%2b14Sp7bruoHnxVEYx6HveYHvhWHge14Y%2bHEUep7rOE4U%2bFEYBJ47P1UQBoFt29YfhzXXsY/y9UoHwSi%2b2EYaXbwK99EBKyvTOkw2EbLSQbczpdNSs4kOMIofsKPnZ8UqlP404IXTMnCUbzw/LXQI%2btmrixLYI1jhpNgACZoVJrk6iA24/%2bydPTurFBpgoQ2DOAUSdJz8YVhzbOuy2X3%2bKntS6WwcZ/P17tOjwoONI3TAbBzndjLl80prP1s7r7SeHGZfFVtJHGUq7e93zy%2bbMMmOTorNrZP8bqYEDblcvb2XrTbQ4UWt/fKyxQqTbK1zeNk8Lbcv6vD2We4wV925qKozK5nP5hXiFO%2b2zI/X23710wKpA5qmJUmSnmCZiD955NpsNuM4bjgcDimKJPp9ghREUde1Pkl0Ot0%2bQY4EQZZkBEYoisYQVBCkHorRDDsckK12ezAYAm2AYRhqSHE8P6SoXq%2bHYT2GZiiK5hgGJ0gc7xE4QZIDBIFH4/GgPxgOKZZhSJLsYThBEDPDIHo4PaR4jiPJPtQFYbhL9oeiIPR6OIogBEmyLMcxDIbgoiiQZJ8k8MGAiqJozTSMPkE0Go1arV4uVer1OtSFMAzrgECtWs2cZhr1BkGQGIqxLMvQtCxPOI4b8aOJLNMMrWkax3Lj8Xg0GkuSyPMjhmZompFEkWM5WZJYjmNZlk7B0BQlSRJF0yN%2bxDBpN45lKYqyLFvXdGE8FkWBplme49JBDCcIAk0zPJtevJDeR4lhOFmWaIYd8RxB9IMgXDMMYyIKBElSgwHLchRFyZLIMrQ%2b0waD/ojnhyTJj0fJqmJtsQfCKA6iJEx3UhyGcRjFfhBFcbJgGCWuH3l%2b5AVxevwYr3d2PzjKfdvZv2q82V8sxoZRnDpgq0NDghwFM2XEEGFLRh0FsyaoNUHtCWbPj4YI6wI0EyD9xrzq/Gmj9I9y1PGsSerAjM4c/vSXw5%2b/Kh%2bttzLfvH729f6TrwoHt042/nr09Kujp3872/q6V3k8wbYEaEOEF9x8a3yAm5/UeeNm3BTAHy2lt6ZNp5bQpLuHeHOfhl6JRIaGjpH6PgOfDIBDrL6L1neJ1r5IZDQ6N6WyK8OcOnjtaNTaVJGS0LYt03Us17GMmW5bhudajm16nvvLZonjJI7eYfyWyTIYR37ozdZURUoSv1ZrnOfyQKfTbgH5YiF3eVmr1QRBWoEY917EcZg6kM5A5CqK2gXBfn%2bAIQiKIigMt9sAgiDVchkAQFmevAN5MtFUpYSMHmcIywkWM7QcB9S5A6PRuFyugF2oUi5XK5VatZa/zNcbjVw2W63V0sDF/4ocPxLH40uE%2bSGP2V64NAfc2dpUnSSRG86TpHeu4mrxpMn2bzGOg0XKvRRGURD61pqu6b458vV%2baFC%2bPnC1fjAbhgYVzIa2SpgT3FIIWyUthZgzNWyFTKmS1pv21P7ytBQ8cPV5HGByub1/FQ/vtS/%2bDea%2bLx/fP9%2b9XTi4y0PPp%2bSe0tud9HZUfFfF91R8T8F3FXxXJdKjhG4vjzsitGkpxJqmTQ2%2bmD%2b4e/pi/eLlg2bm0eXL%2b5m9exd7d9HKz1T7GdV%2bzne3B61n/eZWv7k5aG7RwPNBc5MCnovYvoy/XBIPJGzbVvvzGWBz1aP10vH9/Mv17M7t/P6d7uUj4OK7sxf/vNy/fb57q5N9WHh5J7e3nt27ndu9DVw8LOzfvthfHzR%2bnvS2ReSFhH55bovQhq0Q6Sb2ZkzkjpNATHwxckaJLybuOLT5tCUQA5P1dDowmMBkQpMNTMY36MBkA4NxNcpZGmlbJT1LXMQBB4bRBgABHYTjRu0OhBEUSVKdTrfdQR3X%2byzJ9KchTgsH5jwOxC4MdVutFoIg47EAQd020KEpisB7EIwqimrMdF3TZFlWlIkxm%2bmapuu6bduB7y9OtRTGcfA2Escez/O9Xg9FUJph%2byTZBbsA0BYleTTiu91uq9FkGI5jWKDdYlkWx3vNRksYi7btLDHXiK%2bnEtOppiiK67qrnPy8P5VIfBDsZAvF6fsqFPNJmxc/5gWQxZZYtp/x9RlQp1qrVmu2Wo1Go1ypAq0m0GrBWP%2bHDJ6HxTSbWPblfsQBSZJr5XIH7IIgWCwUOwDQqFSrdWCrTtbIFU2q4%2bsOuJ4XprXR1XxgftgBVU4Sv1Iul6vVarl8enJydPSqUCxrUy0MQ9fzfN8PboZFre9LOzCRxknsqeq01%2bvRFKPpWgr9d0PTNMuyluCALI6S2JvNjJPjQxhGP2e5L57CX9oBTZXj0EkLW28RfhIWA5fgwFSVXVsPwl/q157n2c4nwv6CsEzDtaZvkjme50mC7Pf781Lrm/p9/HlYrMbrxk063xRJEkdXkTh2CZwAgA7YSSOBruur%2beB/B3EcBqmonypxYM3leZgkUVrnDZzAMwPPCn07ClaXoW%2bGvrmmabo3YywZCjTCUVBTghwVCzQ80AlTSmu9pgQbc14ZqS0jS6chQYGjppp4xuYudv5xvnOrmXnYyX6fP7hzvPn3i51bY/jFlNyf9Ham5N6USOX8lNhT55SXKee3r4l6fCHqS5XjbzJ794rH3zUzjwpH35ZOvisdf9vIPMKrT4etLSD3GCr82G9uIuWf4NITtPKTgO7L%2bMFSeShhO/Z0Ieq5fOvsQf31w9LRvcLB3cqrb%2bDCY7L6JLe3nj%2b4BxUeX%2byuV1496F4%2byuzcyu7dKR7dG7Y2JviuiO5I2LK4K8Jbtkqmoj6wxMifJqGexEbsq3GoJ5EeOnISG0lihs4k9pTYU66MyFN8S/JM0bek5VH2jLFvK29qoyAIlqqNehPgWK7ebKWynhw0m61qDbCdhUxLVg1xGomNRRzwEBhqtVoA0GE5DoKhZqtN4DiBEwiCciw74nlJkniOp2lKlmVBEFiW0zV9/jnCKmji2BUEYTgYkATJ86PhcABDMNTtyhNFEAUURbsgyI3GwniMwPBYEPr9fheEJFF2VkHUq/MZmEwUQZLepJOrt1o%2bLuq7IHiRy6vqdHFHA993HMdzPcd0wyB8fz6yOm9oJmqlVKpWqo1KpVquUjSra1Nhot4/xU47wv%2bBqJ9ONQjqdjqdAUlA3e6QYiayPBLE1xCFjd5MS7JiiK/vAUVRDNP0PG82m80Mw7SsL6xOPncPNBv1WqNRLVfOM5nTs9fFclXTtCAIHMd13BvBcZzlKDJ1voQ0XQdBkKJo1/Ms27Jt2zBmvwu6rpumuTQHDNPMZy8Igvyc5b6Ia186Ek9VOfRN3w8WH/q5aY3r4/B9/zcbr5eJFkr/uvEBhDfo886AwPd8R08d8JxZOP8UbXHvfd//6IpfuLpcOLbl2dqbJURRNDn/NmxIUY6zzAThE5M5DEWLxVK73a5WyrIsW5ZlGoZlmpZt/2a94CY1hKs%2bv6vzTZG%2bYZq/oVnMAMOwjWab53gMwVhu1GqD9Vq9DQAIRiSrivhtYWsS%2b0YcuknoxKGdRG4U2HFqO3GUHkPPivxVZOgZ/1tC/wVFBJojbRFjvQAAAABJRU5ErkJggg==' /%3e%3c/svg%3e)
- Easier to move workflows directly from one Galaxy instance to another
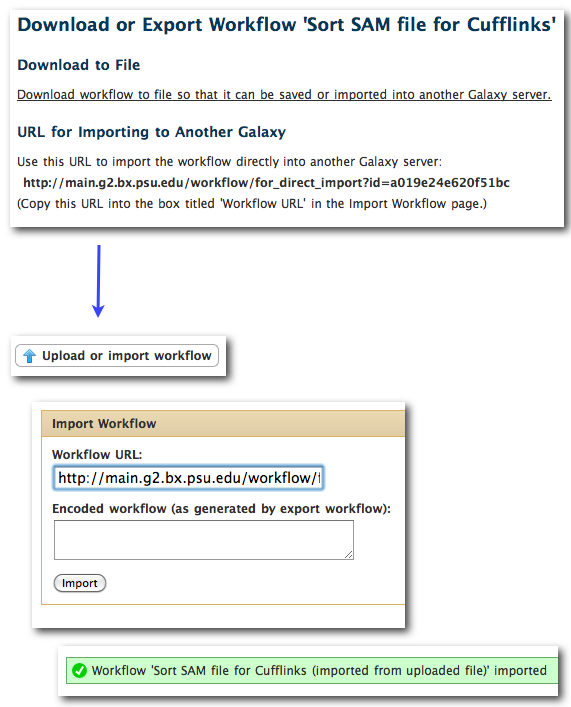
-
Workflow download/export page now provides URL that can be used to directly import a workflow from one instance to another. example: URL import
' width='571' height='707' xlink:href='data:image/png%3bbase64%2ciVBORw0KGgoAAAANSUhEUgAAAEAAAABPCAIAAADUXVRcAAAACXBIWXMAAAsTAAALEwEAmpwYAAATeElEQVRoge07aXPryHHv/6XyIX8g%2bZzPKcdxvLYrsctlx97Ye%2b%2b7V0/H00HxPiU%2bURQlivclHgABggeIGyAIkgCIOzWAnt65m80%2bvT1c6eoienp6jsbM9HQPhnccx7Ft23EcyzINw9B13TAM07RM0wIJXbe8jOdg2w4QcNEwDAtkA7BtGzwMw3ZZju2Ybm0uGI7jgDzDrc10S1mmV9IGhGXbNshwm7Msy2vL4%2bu67jigh7Ztg6bd3JvW79i2ra3XTRgTVwqQc/uxkmVWlDzFWGE2X8qgIsNQFKWLjnXdAF01Qe2yoq5khRNmI4JV1LVlmQwv8jOp0IRdKctxHF3X8vWeKK0cx1ksV2tNx3CKYHiKn40Jmp9LBM3Wugg3XziOQ9AsTvOglGGQNIdOKMdxWvDgstEbTEjbslhBNCxLWq5YQWw2G3ccxyEp6i9P/P/28fZn%2b8nffblz9yD5183Ab%2b/tfbId/vPjwL9/slloo/d9yb/sJiiK%2bv1D35Nw5s8b/j888n2yl/jTV6HTWjf6LPcP//LHj57GP94K/%2bHewX9thn/96dNPt8MfbwX/88Ghoqpf7Eb/%2bOjw0%2b3wh1vBfHsQepb/9Wfbv3tw8M%2b/%2beizw6PEefkP959%2b8MV2KFs5KdZ/8enW7x8Fh1PKl84HnxW/Ojz64JOt39zd/fD%2b/pcHsd/effrfW%2bGnqby0lA8PfXdM01RkpdkftZBRCxnXuig8IlCcbPVH5Xb/ot4tthFxIdehQW9E2qZR6SJXyDiUKVQ6SA0aYlOWEyVWXHQGk2Z/1EYnE5JtoRNGEJsQ1kJG5S4K3hErFFtwrTeYsgI3X45Jutod0LzYRka0IEorBafZK2QEjQhlrXXQcamDmaZFcUKlgxSa/SnNU7yIjPD%2bmICGeOkKRqeM4ziHh4d3vNnmfAuwXXid6bwvsF9r642WLMvy%2bXzfpABYWG/02LIAE6C3xFyl7Fc57uM50wbLwGW%2bKOBl3fy%2bBp7kS6Wupa7bdZleFlDAsixpPs%2bUrqpdFBnh8GBc7yANZAwNJpWr/pQRGj2MF6V8rXOcrxGs8JZ38wOB5Slg27bA80/jZzuRk93EWTB1lslXtxK5e/vxQPoSm9JPgscITufKV4/8qWzlyiv5sg7vTx/7OXyTAt9%2bDfzYwPIUME1TVdXFYuHOWmu9Vi0b5K3Xmrv7WKqiiOLMfeuOaZrAls/nirp23xB4SaqieDPV27C82Wnouu0uGFApqG1tWRag3V3rxa5kWpqmgQ3N8pYK6Nl6vQY7puMosqxpuuFW6/XYAxNsQS9PIYE7SsZj8eSzo3gqdpQ6fhaNxpr1ZjIRi8aS6VQqkUokorHc2XmrXru4KBTPc4FwAsMGJycnnR58FEsc7O6Px%2bNmrV6r1QPhaLfXfZZKFUulcrkci8WCgdDJSbZeq5cLRbjdCUeTyWTi%2bCidjMZisXivCxXPz8OR6FWrGYsmjtOZWDSWSiVKpXKz0TjNnCQioWg83mhdNWvVQDh2cX4ei4RxApjRgwNvH1AUYjrF8QkxnZIEOZ4AICkKx3GCIEiSpK7pKU1R0%2bmUYRiCJBmGwXGcpCiSJMbjMUXTNE1Np/h4PHGJKUVRBDEdjyYkSRJeRSRFUzRBEJPJGLRFkoQrRlHUFMdJwMenxLUszdAkSY7HYxzHx5OxW5oiCAIIT6c8z/%2btrAHDdbm0tSaIkulO/ZWiOg7w2AzXk/HAtqy1prvUtVmwLFM3TM9AAC/IpTRdl1ayDiY3WC03NvvWwXlhRh2HY9nISf5nf7xXbSMjgv7zw73jYnsyxr/wpb7YTz7xxfaSuY1g2p8u/OnBQa7WOb6o7qfO2%2bj4r18dJrKlzw9Sd7eDG6HjyFml1kWfRDKP9uNbofTT2EkkW9EM6/2OgGma67U6pfkxQU8oTtX08ZRiZtJaVUlu1kFG3T42oTiCESh%2bXm1BDWg4l5YDnFrIyhAnSYbvj0ma5QcTAqcFRQWeDIxNkNF0MCG7GP7mALwYE%2bsted9%2bV/lbWQOWZc3n80EfhhGU44VGpQL3UZZhS6VCq9tbSBIEwRzL9rrdXg9qt64mE3w4HJULBZpmB0gfhuBWvV4o5Kv11ngCDEu9Wu9eXbXbXRiCZwLfqjcQdMBwHAT1URSt1%2brtVpOi6MFw2G02oD6CDlCW44YDDOr3YajX7Xa9xfba8HyTAktJGqAI3IeFmYjCkNtjrtttoxg2m80g0I/ZoA8XLi%2bbzRbPz0iSbtVqNMP0ep0eBKN9ZIihrVZ7OMYJkoR7ENzr9hEU6vVmAo/2oD4ymBLk1VUHRdB%2bH2k3axzHUzTdaTY63V6j2aBoBhtgKDZEQWHk/zYCuq6DwGopi7P5crWyTFPTtNVyqRvmcrlSVNU0DYblVisQlBm6rqoKkNF1cT63bUtVVRCpyYrA8zRNy7Ji2Q7HcfP53I2/lsKMX6vrtaJouiErsqZpBtj7FY5laZZfybLjODzH8oJIEuRKlmczgWFob5OZTHCoB5MkQTOMJEmvDcMLBRzHGQ3Qrc29jc3Ns7PccIjFoslIOBpOJk%2beHe3t7R6lTw/3D7K580Dg0H9wkMmch0Oh7d29fP4il80%2b3d1PHyVjkcjRUXJ/35c8PqlXa9tb2xAMn%2bXOAgFfJBxNpxLNdi%2bZPvYd7u3u7ccjkWKhFPCHTjKZWqW0u/P0olgJBf2hSPj0LBeNhgIBfzwWi8djoWC8Uiyljo79gcC1HX9NAdchMUzrOioHbgyI0K%2b9%2bfV6vVwuvKjAsmxNcx0kENpf%2bzOeVXEDXxCPu%2b4TAMDRtLWmuVXahmHKiuLtHrYbIdyA/dIsd4P6NyIQG4TqXzuFbBuM4OXFeaFQSh8fTyZ4pVTOpI9hdFgt5gPRiCRJvVbr%2bOgIgpHyZfE4mRqiSCJ5XK5UMGyYTh2dnZ5mTnPT8SgUDJ9kzi5zuePjdCZzclkoYUg/GArVW015KTUqNQTqhsOJDoQ2arVsJpvNpM9zl1APjkfDmUwmd3oajyfPcrlKtT4ZDWOR%2bNHxs2arSZLUW0Oyl7xRRZHm85ko4uPxcrlUFFUUZ8vlarlYiPP5er1WZWUmzJYrWV7JM0FYqyrH8/O5pCrq3IXZTDRNS5gJ87m4kBbL5UoURUEQFFkW5/OVvLIsc7VYqKo6m4nLlawoCs9yLE0tFktFVubibD6XFhIoORME97nCxxMEGYiiqLiewd/mPrC/vw/caRiGI9cQjkai0Ug0EolEo%2bDXI57Tb8l6XjASi8WumQBfFYgCvxnU7CavKwxH3qzkBqLRaNgFj469DSKRSDqdBudCsixzHMfzvPf7MggCwDfYtwBvtvUacC54D/5rQFVVoMBbwTO6OOGQ9IvkjxDueI83/VUTeMp2q232YNP1pu0fITg3CrwJrk132j0LxTyT7/w44c43K9BsWyj2Itz%2b6SnQuLIGw/9X4AccgVLVhPo/5RH4aseIJNzw/H1Ftu8K/8s%2bIMzsxeJHb4VsN5h28Ro8K/sm8TJcn6e/lPXDKGB7R5mGDlDXteewXoNTyfV6rWngM90N36MBG9DAEXRlAH82m3mRwPeqgGVZiqIAT1lRFUXRgRa6e3oKwkVwLquq3tGsN0IgJnQDGl3XFVVZrlYerarq6enpbDb7nj8g3DEMU5MXHUZ9UJtLHBWLJUPhcCyePNjfbbZbZ6eZwWh6fnGRSMUTiXgkEms2GvV6PRJLHSfj0aA/Eo5Go4nWVWe9XkciEY7jvm8FTNNcqzIlyoXJwtKUPoKg2HA8GkEQJMxmU3yyWK44jh%2bNMBiGcHw6F0WGZlAUIwmCIgiGpoejEUWz6/U6FovxPP99K2CZ5mq1mgm8JAC/VZIkaT6XJGm1AsHRXJoLPD9zIyZpsQBJQRBFcbGQRHEuzueiKEqSJIoix3HBYPAHUMBxHAzDwpFIIBgKhUJBFwLBQCAQAEQAsK%2bZLseT8WhXMuiRgUCgUCh8/8HdHecnDnduKNeYWrpp66ZtPMfX6JeTX4POd0QDnOt8dwXAqZtp6aqozMf6itCWU2051ZfT9QI3ZEJ3aWNFeFmqNFHmt4lraVIt5%2bdz6Tusn2sFwI0U05HZq3F1s5t/MqjsDCo7cHFzUN5uZR/CxS2o8KRx%2brB19mhQ2Zm2Dpiu/xaR6x36tr%2bkGe5dFVC4q8azz7/67JePP/n5w49%2b/uiTX55HP/7yw589%2bOgX23c/2Ln36527v/riw3/Nxj5jur5JYxdv7t0KEq3dg%2b0vaIZ/NwUsZ8l0iHaQQ47wdpjoRtl%2bkoYSNBSnoTjTT7L9JAMnaDhB9eK3iwwU9z29/04jYLtfeQ11ZlsvDlC9o1K30mtfz3FscJAKbix9F7Rt03kDbQtY3lg0QlHUOymgabpjrgSODvr2q%2bVi7uI8kYhe5E7LxQJF0blMNuA/ZEnCuW2w3R7HYrF3VkA3bH0hSfPTYvO03MpWO6eVq8xlPZUrtzAqW%2b2kL2uFFjziVvhMmQjymFsO2cWYX10T3HI6k8fcYsKvJi5zIsjTmTIVlRG3BExBUcE3d3Dy7byEtntYHYtFb0EBR5emMznXwZsD8gqjhuyyT8wgnG8P6f5U6E24AbOAJlyxjZU6WHvIIOS8PaQ7IxYhxd6EvbxCezjfREmXyVR7WLbWy5Ta0FTojuhMFerTyze7aN/iCDiGNOaXhVIxEozubzzoXjXw0bB0cXqZTcUCvtOjFIzAkYAvd3LSKGQTkdAA7Uf9hxfZk2almj89frr1JH%2babjdq8VAglUicRPynJ6lkMhHZ38mmU/VyfsDK71kBXaLE1QUi1IdCtoEV%2b3SxT5dRrj4U8hBRGfAllAlmG5cwXRmwJYTOQ0S2NSr06YsuUUKYYp/OtcclhCmjXKFPVzG%2bPppVMT7fnVZQ5hxiRxy49%2bcGG88DPcu6NQVcKySCy48378b9NW2AXvImsjdt8OHkta8OHm07jttDIGxYLwRM8NXnLT2wb20EXCvEUMTFWbZcLJeLxVQicX5%2bXq9W%2bzBcKZfzF%2bfPUsmj5FGlVBqPhlfNerlcZqjpWea0UCrXqpVqpZLP5S7zF5VSuXiRhyB4OEBL%2bfPLi3yxWOx0eyiCFEuli4v8s/RxJBQtlyuly0sExRzHiUZvYRHrtg78/G6ne9Vqt5qtWrVWrddGw9EIw5qtdq8LQb0e3IMb9RpBkDDUrdcbkxF2fPTsqgd1XWg1GrVabTgcwt1OD%2bpPJuNKsVAslNrdLkHR%2bHjUbF11uxAEQWgf6XU7vW6XpNnbUQBcZ13PNW29XC41TVXdUHitqrIMvggBSgVBs0trsgtrVV2tVpqmqYri5YJSz4soCpBR12vAUtXVcgmKuPRyubzpqH2La8DSJFWR12v3NtZL8/stty1f5nxTk68ct9ycviyXS2%2bbt293ET9XAHxLfa%2bwWq08J8V5HyPgfhV8cch1uwCuamuaFzrfvgLuGlAX0kKW5dX7AVmWBUF4XwoY7j5wc1Z4c1HQeZ9g3/YIrMdDrNvp9WEIAoYTpijqdnV47SDVvkVXwtIkWV5dNerP0tlo0Hd4cJDNXgywge5ekbjt73O37Y3q3hSyTVlZg781rGTvzw2qe8f1JzOFdFmw9IWpSZYmmS5ammQooqHO3xPq6tzWl5FwgKLod1AAWDfdvRqsm4b2Glqm/v7QNDTHsaLvGlK65tn5IcB2exyJvMMa8MqIophOp/1%2bf/B7h0AgkEwmZff62XccAe8MYrFYiC7wAs8JHCdwvMB/D8jx3HK1vP5v17fGr71q8Mo7sN5Az/rd0G8lXpZ5s5SH4AaGc5O0DdvWX6ClWS8nHf31XMdyKIKSJOl1BSzb0jUdV6aw2kdUBDUGmI0NzIGLKGZhfa2P6AhmYaiBIlp/YIGsPiAwVEcRAwXJNZAZmAPUQAcWhlmAgxooZmOgNmswsAZDG1SFAgGQBPJuVRiQB2KIjnjFEQ1BDOSmoNcfzMYO0/4BMniLAsbaqMm1vx/%2b3W9n/xHqBbaqW5FhcKOwsQ/5wsNImj/ea%2b/7kUBsEg0PQ5uVrd3WbpxIPCk%2bjhMJP3wYGkVTTDKKRzcr208KG77O/iESzEiZAOoPIv5D2B/CggHEv13fPaLiPugwOAgGBsE0n/ajgcN%2bIDoK7bf3dloHcTy223p60NsPD8MJInmIBMOjcHAQPIR8Bz3f6Sr7NLuDwujbFSguiv80/sdfsb/0dw/uXdzfbmxuVDY2G1v3c/dTwvEBfPAg//Cw7/MPDh8VHm81NmNU4vHlAx/i80F7G/XtBJfYam4%2baezstra/Kjx6VHkSJWO73d2NwoNPjz/dqGxsNzY/O7mbICP70N7j0qPtq5393t7nJ59vNXcenN%2b9n7t/7/yhH/MfQPtfVR6HJqEQFnhQeHQ/f%2b9J9clG%2bfHDy0dH8%2bOt0y0UftsI6JouWAIJbjrRjMNyDks7DOswDPhlPT4LmDTlUKzDMg5DOiTrcLRDu0VAknEJtyDLuhyvNk/eq5Z0KMatinEYChDXTbBuLuVQXhGPYN3armuwGdbhgs%2bCCIy8ZRGvViuapPExPp1Mp6NXcTglxgQxJqajKTEG6PE9zg2%2bkhy%2bynGT09EUH%2bKviI2fZw3x6RDH3YZuanutfnyIsyQbj8ZR9NUp5Nmf2Wx2kjmJxWPxRPxHi9FY9LJwqSjK/wCOaRqV3JaLrQAAAABJRU5ErkJggg==' /%3e%3c/svg%3e)
- New Parameter settings for global application or specific actions
- Workflow parameters are a new feature we've added to simplify reuse of workflows, and to allow for easier variation of parameters when re-running a workflow.
- Instead of filling in explicit values when building a workflow, you can now use flexible parameters. To specify a workflow parameter, simply use a tag like ${my_variable_name} in any tool input field or in a rename dataset action field.
-
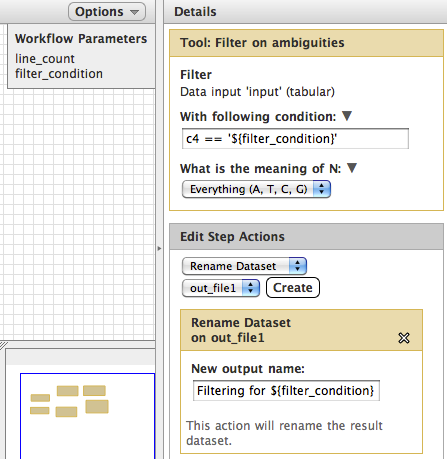
The workflow shown below has two parameters, as shown in the Workflow Parameters display in the top right of the editor window. You can see the ${filter_condition} parameter in the right panel in both the tool input and the rename action.
- Note that while this ${filter_condition} is only used in a single step in this simple demo workflow, variables can be used across steps. example: Parameters
' width='447' height='459' xlink:href='data:image/png%3bbase64%2ciVBORw0KGgoAAAANSUhEUgAAAEAAAABCCAIAAABow0eCAAAACXBIWXMAAAsTAAALEwEAmpwYAAAT2ElEQVRogb1a93fbSJL2P3s/3A97e3tvw9tws7c3NzuzO/Y4K8uSKIlKthIlKgcrMIg5gyQIIkcmEDkQvNeAJGucRrblrfc9sgA0SHxd1VXdXbjX%2bYCIoshxfB2pQVCZ4ziCJEiKYjmOcYUgCNIVnudJkqQoqtNuEwQuybIkdVVV5ThOkiRRFDtfWe4xHxCaplEUu4iGhweejIxPTE36xscnfZMTw8MTp2chuFaLhMMYhtVqNcJlUy4Vnz1%2bPDjwdGp61j8zMTYx7puaomiWZb6u3Cu5UnTlWikWChAEZTKZ8/OzpcW58YmpQGBjaWlpZXn55dLy5mYwlUqFw%2bFkMplKpTAMK5fL6VRqK7gVCATW1zdfvVraOzgIrAey%2bYL7m18R9yAIKpVKdRSt1WrVarVWqyEIgtbRarVaqVSgchnD8TqCVOEqXAMC12AYhiEIKpfLlUqlUChgGFav10kCY2icYUiaJhiGokicZUgCr%2bMYgrnAMVdH3yhXJ8HhNUADHCHw%2bq1AYPdarRZN0zRFsBwvyRLH8TRF1mqIrCiKAhy6DEHNZltypd3udLtSu92SJKnb7cqy3Gg0CIJsNgSZPWmiQZHck5kDmdmX6P0OsSMzBwpzaPBHKnck0XsKe6jzRxK1p7qKzh%2bq3KHGHSosaOYq4FOi9jv4rkh8HHsdLCixoXuCIOAY9vj%2b9z8%2b%2bGls/MXw2NjI0LPBwSej474nDx%2bEoin/5IuR0bHZmcmR0Ynh4WfDo2NDz58ODY3MTE/555dQFKMoSuDZJrpeS0wE5r6L7D2N7j873ngIJyaWp7/dW/nhMPAodvC0HPdlTwZPgo9rKV/yaPAs%2bGh7%2bcfDtX%2bebT/JnI1e7D7aW/kxtP3k4mCgmvDx1SUGWmTLH8ESXZoVkM17PC8wDO36MQRBUA0BLoJhaCFfKBaLJEnxHJfP50rFQjKVqlbLxWIxl81l0hnQvlwmSYokqYbAtbAgAy0drdwff/yHyeH/Xpr5vnIxNf/i27WF754/%2bMPM6DfF%2bOzK5F9HB/6SD42OPv7j7PA3I8%2b%2bmR3%2b84Pv/mPw4W9nRv7kH//bzNBfxgf%2bHD4YEbEAX10W4JUPY5WvLLaw3XueD5A4znOcIDTEjsjzvMALYrfLsjTDsK1WWxRFsduVJMmNrmLXlU6nI3W7IMISZEPgBPglXXjBQdM8NM1B02zJxxSnhMoMB00LZXCGg3xsyceXZ9jSFFPyCeUZHpoWKjO1%2bBCSGuPdNu6njy1OMb8MH5UbFWprwIUIgggsLm5vr0/6ZiEIWltZCawuvlzeXJ2bOj6NK4rcBNJqviPtdptlWYIkBZ7RxKqhcpbGmSqrS7Qm0YbC6DJjeFAuocu0roAz%2bhUsjbNU1mumy/TNxh%2bHLlNKuwwIUBRZrUBIvY4TZLFYcFMUgaI4z7E4TtA002g0Ou02IPFzuSJACTxt642e09c0w%2br1Haf/rxFDYYEL1Wq14LJ/anJ%2bamzw6CSMIPXNwOr21ub8wuLywvTw0ORGYHl6du7k5Ljrhp1W69Ia7XabcwnwLNnvi0i5suB/MftqddY3vRrYYFm23%2b/3erbj9AB69iWcHjjZs91P9zxo4H727L7Tuz0MmQIEKIriGLIGI5VyqY6guWyuXIZgGC6XK9VKGYZhBK6WihBJEh1RbN8QURR5jiNJSuContlUZK3T5AiayWYz6WzeNI2vbgGZBgQQBAGpgAFDlnKFuZ4BXCksy1A0ReA4cUNIkkQQBMNwgacdsyXLSgkqURSYICmy3GoIGE41eI7lQFSgaYrE6wzLN3gOxTBB4DG0jiL1OoLwQoPBMYpmSQw3TPvTCOi6riiKJEmyDPKW7IokSd5JN2Fd6t4lkN8UxdNVVe10OjhO8BzVtzs8x29vB5cW5g92d8LhGAGXFhZXd7derW3uwtUqw7Knu6sra1trK0uh8EWn1Q6Fz17NL%2b7t7K8sr0WOdoJb%2b4mLmKTo/X7fud0wuiRguGK64umGYViW9V79ZjPLshRFJghS4GlLF67/2HGl3%2b97jft9x7Z7vZ7Td3qaqnhNer2eaVrgpNe%2bB8aJaXya1wEC189kueLp3uH1Jdu2r5/7ZhvbthVF8QjYuuCAIdvr2Zc%2b0AMj0rFt%2b5bd6YnjOO1WS9W0/hdawDTN60vXFtB1/S0reRbgObLviBRBjjx/OP9ybcY3kchkzw%2b2FxZezc%2bOb%2b/srK6sHR3sBYKbq8vz0VgGReHA2vrO1rZ/2r%2bwtLi8sra5EQisrm4EDx3beHT/HxepgtsFvX8RARCFjKah6xeRcDafTyYTdQxjSSwaieZy6XQqGToLxePxQjG3tbGyc3DGstTx4XEqmTo%2bPI4n45ls9iIaiYTCyWTOMPRms2kYhutWb8tbxgQENE3TXbl%2bxOvD60seGV3XNU272cY0TVm%2bHAO23rBtRxQ7YMbRbrdb7XZHlGVJEAShIbRarUaj0e1Kuq63mg1BaCiqKkmyqqmgndjpSpKqqc0mmNdwHAfWU%2b8IRVE8z79NwOvsa%2be%2bOSRuXnq32ZUFlJsEaIJ4fXxYrlQEnoOgEoaTotg53glube8GFubD4eju3iHJsDzHhk5Psqn40dHpUXD9PJzIxCJn59HXezvtrvKRKKQoyk2/uuNB3HP69VotHA4zLMMyNARBSB3r9/sXodPzUCQbiybjiZXlAEFR%2bUJhfyeYSycO9o/DR/uJRCZ6evD6/CIaDlnOVRR7R/r9vizLbxO4WxfSNc1xHNuyvL/pOY5hGLbds2zLcS77VXZntZfd7DgguvbBXMINu5e95v34u6Lr%2bk3j3N0gvsoDnzePc9ybrnr5E%2bTOLAAysdVutzvZ5EUklmRohqaZjthFUdww9EI%2bU0UwsIRot2AY5gUBrKJoGqnDDEMLjYZpmnClBFVrVaiEUYzYEqamfB1J/cWUDAh47u45tKd4g8Hz%2b%2btL7zbzdFVVLwnYHZ5lB588mJpfnpuZnFt6RVDM9NQsiuJrr/yjPv/B/n46lRsfHJqdezX5Ymx1M7i0NLcRePXs2fM6ik6NDcA4EQysrmwELVNfW10zwGj4V1kAuJAGXEjTdNu2dR3caFu26Q4GywL8NU3rOWCQqKra7Yq6YaiqqusagtQUReu0FUW2LLeXbu9JdzgGQCLTdBOr10JnZ7kcWE9XqpXQ%2bXnkPFrI54ulcg2unp6ckhQdDYVrNTQZi9VRLJ/LpuKxKi74gtBDfzycb3pD376aj3x9C0iSR8Axm7KiwxVoZ2PD759bXFzZDKy/GJtc8K2c7O0OjEzs7u1PT0yELuKFTDaTSh/sHSZT6fOTk%2bXFuTQi/HOz%2bpup%2bE6m5RK4pQHuOA%2bAJSVFUThBUBRNkjRBkAzICAAIUsdQjOM4FEVJksQwsK6A4RpFUWgdxQgqh9FJGG%2b0u58UyQyZuTMXslSwgAR/DuCui0FYBAvFy0Mwh7a9by9mus8A9DfK5b23hS6RX05AIQii2RBsnfc2C0yV/QwYHj7pdoU2Vf5OohDebDZ7Tt8wzGu4ORUoumEapmVcHlou3DamZYIw9UWwbedOJnNEo9EoFguJZDKXy%2bUL%2bVw%2bn06nc7lcoVAoFgq5XDabyebzhXw%2bn8vn3M3vQj6XS36BpFKpWCwGQdDdEBB4vlSCNtfXRkeGnj4dmHgxsbW1MfB8wO%2bfm5tfCG7tLMzNTk/PzPtnfVPTfr9/YXF%2bbT0Qj8djnyvxeDwSiRQKhTtwIbC5KwilUmk7uDn4/MkPPzwYHRre3NwYeP700cMH//O/376YnHm56H/48NHQwODA84H79%2b%2bPjQ0PDg3H4omLiy8iUCwWv2gQm6apqmo6naZpulQqJZPJbBa4TaFQyGQy%2bXy%2bVCwC78kC/ykWgQsBpyoWC8ViPg/2iHO5dDabSX26pNPpeDwOXOjLoxCCICzLSpJkWZa3%2b9IFW8CgOgZ2gkVwBJZpIjjsdNytYneDjGPbNCU2G90bqeUTxDAMUCP78kTmrfQ8tgxNJWPRKowQOEFRVL1ed3fA8Hg0iqIohuM0yFxIHUHFTisGNaYC6fn9eqdrfN5MXJKkLx0DEphKEBzH6bouKwoCV1YWfGMvfIuzK4MDY9NTvkX/bPjiYnJocGRkZHzCNzk%2bPPZibP/gRFe6O0XpT/Pp7zcpvg02UT55NeARuJNExnGcqqqO46AoShAkjuM4gWM4DgqyQCdQFMUxApRzSDKTyYL4cxHdO4suH50HTy5QFG21Wp/B4Q4s4CYyQEDTNMuy3PIsRVNgQVOrVvLZLE5SWB2tYM2XO9CrvRpU4%2btIJZNOQeUKReA8TaK1KkXRxWJRVT%2b2nP8ggbtY0AAChmHIsgzD8OHu1qx/eXl57XB7Z212Yu7l2tlxKIUrv/fF/jBb2g4jLFWPRi9i0dDi0uKcb2pmemZvd58kyU6n8zkE7tACtmXV4EqxkM9lM6lkPJNOI7VKLpsu5jPZCrKTqW7FKxTXgCulaqVcrVaQWqVYAFv51Uq5WCwYhuatBW6DrzIGLhfmV9PL/vsUMAc27U5X7XTVdkftyrro6ob5C7uIX98CumnpzSZ%2bqDYuFD7SZUMSG%2b6yYe%2bzy4RV4ULiIl0mJHNA1xoxtQHOSFxE5iIiE2pT5x36FxFqk68lIeP2igMI3MWChuA4Vjd6Whdli6OZ14O58%2bF6ehJOvMCzU3BiHE1P4Zmp%2bMGTSmwcjo8jqUk0MwknxqvxcRbyM6VZpjTLlvwsdAuU5%2bjCZKO%2b4T5/7w5diNUNWxXxLrYwPfjXb//0b89/%2buOzv//66f3fP/6//3x8/3enwWcjD/7rwfe/fvrjb4Yf/e7Jj799%2bO2/ry0%2baNbXueoyD6/cBq36WgtdZ8sLLWzPc1dAwOt4L%2bx4EeatKGRd7ixc2udmm16vp6oqSZIeAV3CmeIUV11Bs348v4BmZrHcfD3tRzKz2fOxYmSinpktRidq6ZlaagbL%2bsniAl99dXtU4j44OcNCc01s940Frou%2b75ZQvZNeNfJa966%2bqVJyHI7jHgGti7dr/sD8Px7//Vehg6GNhR%2be/vDr2fFvsucvxp//8advfzU//teVue85eK1RW/1oFX7lXTRrKwOP/zY58kMTXmxeWsAlEIvF3FzpZcxfkOs2INfiOEmStVoNwzD%2bcgxgTH64FBlJHz%2bup1%2bUwkMXez/lzp7HDx5VE2OF8%2bfF8CAUHWaK02zpcxDa/il28IQpjDeQzTcEIAjCP1cIgrgiwGm6ZapsizjWWlmtmZa4hNJIGZ2s0kjJQlLiwaGHLhvvMPEOE/MgsokudyvorbTeyohMVORib6IQgiDtNnCZZvOT0Wp1GAa8K%2bEmMsM2RcvovhP370ycq29DYd/kARRFwRafoZrmJ8OyNEXpMAzFsqymm6bW6veVxEVkcysYj8Vy2QwvNLLJWDqdSsSjmXQyEY/D1TKJY%2bdnkVw2EYpELiIhjm/8rKD/PvxsN6VnGzL1hgCOE/1%2bv9Xs1%2bA%2bWn%2bDOuJ4eEu/CRzrFwtGpcIIAqtppqW3%2b32lDJX8c7Pr6%2bvBzW2CpIJrr9YDgc3A2tLi7Kvl5Vw2g6P1qfHpzeDq3vHrRf90FaE/bQrkOIZMv02gAvXFTl/XHU3tAWh907qErvdN8xKa9gaq2u/1%2bnXEiMWYVotVNdM22rbZNU0wthRF1nRd7HQkWdE0rd3utNptrzDebDZlt3KuKCoombVaLMuASlqj0Wo2ms2mqzQF8FJGm2GYtyd57yVQg/s3a8yOYwn4EYvs8uiBLiF0NUiWNxh4q2cp197o/SDLGMnEJQHLJaCoSiIaPTk5S6cSMFwtFbLpdJ6mabhSjYTDdRQnSSIaOdnb34%2bcn8TiuXqtKooSSeC1agUq1zKJeCqZTCYyGFbPZ1N1DOtK8i9boAY7ptmXW2WBuGgzGUvvlMLP0sc/5s8etaloKTJUCA2UL0ZsU3qLAHODALCA0dV1LZtKbgf3AsuLK6ubicj5y%2bXg2pI/X4T3tzb2j0KtBr88PxncPY6cHY2Ozx/vb/PN9uvj4/2t1a3ds5fTvuOj14vzKyevj3bXlwtFSNGMn0WF9xOoAgJ8/aCW9GG5xatgYvf7vb5jN8kwU9tn4B2lg3o/8UECZlfXTct6s2wAimmQBG6Aw8sJi6ppOqid2aZp2JZFUbQodkE1zbINXbue03i7HtpbVfuPWKBNRxl4v0lGTbXBoacMcizg4Z6llCLD8d3vLra%2bYZEjL4N8gEDH0jvghYOr1wu8YvW17m6dg3r11fsRjrcDfFUjA6X5ywn5O/XJWxHg6of1/DKDHCvtWv70p/TRP0uhp7Ypd/i8gIcE7EwTyY9aoGMZ4tfLA580iJ2erbkAL7%2b8V94moBqO1e00SagIURQrdsReryeK3YbANxuNZqvdFbuaqvEch9dhWdW7YkdoNDvttiR1WZZtt9q6blA4SjNgdSqJYrPRUDVNEBoMzdyewPtM5vrMFZyPEOiZoio3YKgcjybTqUyrKRwevU7EY/lM%2buQsDFerGIru7x3kkrGmKJey6dOzaPz8NBq7ODk5DZ%2besnwjn0ru7xySFIVUy6fHr7O53Nl5KJ/O3J7AJ8g7BMyeKeqy0O0qGniJStYNQ1Hkble6HjZv3e%2bJ7ZXEPe93x4RX9Pbe3HHfI/p5n34lAoqq9x2FrOf3dnYOD8PHh4ehaPgsFIrHs5ftb9z1oX0453Z/bMjMV7CAG0Y1hVNVRZa6iizKkqhpsqp0e7bpONadoWfqMnjmO3ch3bE1XaZsXbA03tJ4W%2bfBS60abyqscXfQZdp0a9L9PphO/z/Rtg6V1wI4CQAAAABJRU5ErkJggg==' /%3e%3c/svg%3e)
-
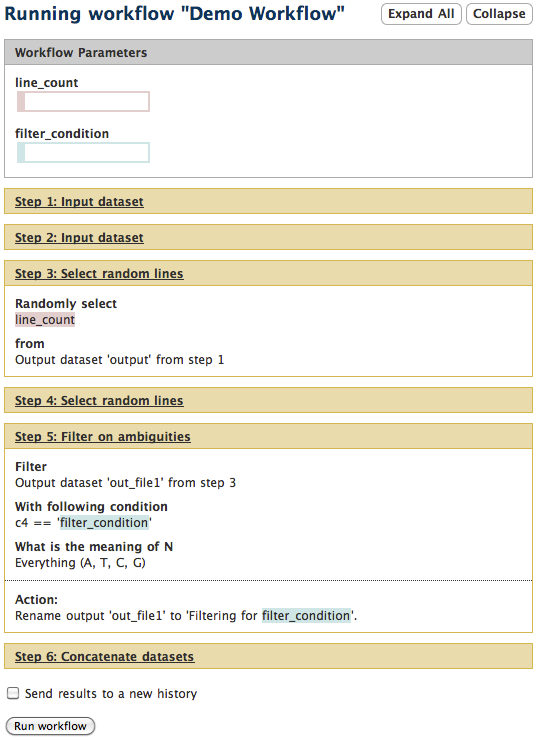
As the inputs are filled in the Workflow Parameters box, seen in the runtime example below, the new values will be reflected in all workflow steps and will be used when the workflow is executed.
example: Runtime display
' width='537' height='740' xlink:href='data:image/png%3bbase64%2ciVBORw0KGgoAAAANSUhEUgAAAEAAAABYCAIAAADKjmZ/AAAACXBIWXMAAAsTAAALEwEAmpwYAAAS8ElEQVR4nNVbB3PbWJLmn7yfcHVXdbWzE%2bzbCbUzezf2OIxsy8rBki1ZmSIVSAXmIFKkGERKzASYEwACBEAixyuQkkb2eKp2b2dEbddXr7obDySa6PfY/V4/HdJGL3NlVVUFUeR4geUEGMF5UeQFUZIkSVZkRREEQZaVJowiGBG5zFzmq6KkyLKsqmql1vSfp2KZEsXwkiT3aFa7UVa63V6uWGvAmKyoOEHCbSIDlL2hlKIogigqshIIJ2mWV1VVURRVVVmWxTCMYVlBEDiOoyiKJLskQaAYSpAkwzA43un2KAzFaJoWBIHtE8MwOof39N//Or5ktEy83331dufFovHl3O7rxd1HE6trB/58ofT96/ejC8ax5f2RBf2jybXFNdP3o8tj7/Z%2bGF16t%2bOiaHbv2P14evM/f5icWDL9OLnx6t3u2MpxuVr/tz89/mF87cmccXpp939eb7xc2n82tT26YHi1sP3Xsc1oApAk7SfoP7/KcZzf6z027Zn3zUaDyag3hsPhxEVky2gEyg2a6qWSlw6bTb%2b157Q7jNu7tmNzKBJjWE5Hdnv5cj2SzOdLdbDSBCvNDFivN9v7xyd665ksieEEkC/WEvlKA0YzYAVGOoVKI5kvJ/PlXKmpqmoHJ1NAJQNWgXKzWIVyxXoiX%2bFFKVuoFevQeQpsQGi%2bWC/W4RaMJXPlDFi9yJYkWen/%2bhqpqkrTdLuNVMqlfB6oVmqNegOGEYrqwQhCkN1er0fgnWqlAsNIvVbPZXPNZqPVgmma0an3g5RrSwbSQNZcWJYHennQXosD61VV1ZEkWalWkXYbguEWBCEwAsMIBEGNZhOCYQRBmq1WqwVBEAxdU7PZrF1RvdFo1Ov16rV40zQajT43uKQxt8SrDp8k7ROvqdlsftQ2Glpbr9cbjWa9Xu90Orp6ve522tfX13f39sxm08amfmNj/fDAbLPb9/f2DMado6NDs9m8v79vNptNJtPBgdnhdIXD4dDvTWe/TZ%2b8enp6CgCArtFsOhz2Z08eT01OLCwuzMzNzc3Nvhx58fbtO8P29svR1wvzM%2bNT03PzM5MTk%2bPjE/Pzs/ptQyQaHXzuECkQCGgGwDCcTmeqlWoeAEAQLIAFoM8AQL5QLBQKhb4EFotFEAQKGgsCAJC/B5TL5Wq1mo5lWUmS1H81UvojmKIoHcfxdA%2b3Wu2mg6Nsvgjk0g679cTnt9qOWwh20/X%2bGsCyLM%2bxQDoVi55nkulsIhGPhKOhUDpxgcKoxLE8zQjMh2DZoVul3DZAFPhOsSALohYe9AOE606yLEkfQZFlnqZFXosC7osBAs%2b3Uimm25UEQeQ4geMElhU5TuR56VeQBYHr9e6RAQzDiKIo0DTPMHzfPQSW/QTPceIt3CMXYhjmX3sW4nmB7GB7u3sWqyUQCMJQK5NO%2b7z%2bE7dnz7jn952cnZ2FQyGhHzne11mIZROZTDAS9oci%2bVIhFI2enJ4FQ2GvP3CRSpyGQ5F4rMvxrCAwfbCCcI9ciGVYQRTLJCn055/fei5JlgeQFYXheWHYXqd88AYEIQvDNM9LsizKsihJH%2bP66QcG0DzPi%2bI9MkCUpB7Pa74hin8n7pEL0TTFsowsiZIo/P0QBH7oEEWhS5I6SVI4XmY5ieVElpM4XkNfvMIHYp/nBJm/HxAlWccSAF4%2bpGEvA3toyN1tOMmGk2q5ByLVcncbrhuehtw07CbrDqI2dDjxqpVGL3RUOwaGxg1LP1gMT72Hr849U%2beu8RPTyLHxqXPvudM0kvBOGpZ%2bONb/5Nof8ZpHjvU/AedL7fxGK7MOZYeIjVb6fafm1NFoDAiOLc98PTf6xfzYQ/vuz4cbP%2b6v/e/s2Jczr754P/etY%2bfJxPPP3o4/WJx4%2bH72L4vjD7KhRQzYhDJrcHZ9eNiA0u/xmksnMG0KS4l0UaKLElXgSIAlAaEHynRBogsiBXJdQKILElUQKa2V6AKD56hOjh42KCzDUQ2dwKJ0Jyv0ihwJsgTAkiB3DZYAOAJkCJDvFvqihoGewYcPupPjqZauB0dKkddn1lfp0ykwMgeEZjKB6Yx/MhWYzoVmMsHp0vl81PEqcTKRPZsBzmaSvsnyxSKUWWqlh4vlZnKhU7HpGOwyH5x6/fTPI4//NDbyYGf1sX7px42Fvz1/8tnIT59Nj/3Fon/y43f/Mfb08/GRr2ZGH4w%2b%2bXMqtIyXduD8NgIYhgcjnNsi6l4dSxbRkgUpOaGivQnaoIKtCVgHYgu0wQU7VLRD/bYF2qCiHS45OlUXXnNfoe7G655hwNupOSk0oZMlTuJJRaZksSsLpCx2FamniN0BoynFriYKWjvQSDwpCRpEjtD4YYEjZJHR9ZDz%2buVc9fI9kt/AwM1WeqWRXqmnVuDsWjO9iuTXW5lVKLuK5Nbb%2bfVGegXJrSP59UZyuZleRcENJLcOZdeGgfVWelmbRplOAgjNjD37YvLFw8Xpvx4bXuyuPttZ%2b2ni%2beevn381M/qNzfjiSP/z7OiDubGHo88%2bn/j5wcrCo92Vx5OjX0%2b/%2bnr93Y%2btvLFd2EHAO8YunN8mGic6Cr2oXr6LuucD1kmfZfLSv3jumY/73gQd0xHX7Kl16sK3kDhdCNgmw67ZoH067JyJ%2bxYTp4vn3vkz52zEMw8DBgQ03jl24LxeM4CnIYFF%2b5mMpKqyqop9SKoqqArbZ0SNVyVVoa9BqTKltX1R5MlhgBCFHtet6mSBpHsdny9UKlWSyWQkGsvlC/F4DCiWyS5VLpcTiWQ6C4BgSZT7exLXWVt/hf43M7g7yAhUVRVoSCfxOMfgh6ZDr9NusRyY901Ol8vvdwfPzmq1hvXIbLMd7%2b%2bbA6cB8SoLU%2b4H5GsDBILtomdHRyG7o5G4zAYCl15v0uMKHls4Trg29R6S8oEBdr3esr3tOzB7TCab0ejeMXpNBwzD9RcXtbT4/q0dKVcGiGxHlv7hdcKh58TqjQGKSJI4cmAynfr8FosTgtuDvbQBKdoCr9beZq6e/ka4ujwY0cqduxCHCxwN5IFcNptJAzRFf%2bIH/lDsmyYN%2b80oN7MQwVCYzxdIJC%2bj4bPzaCSVSsViUfuh2XNyGoqET1zOeqMVDgUDwbPAyUkoHL6Ix8ulosvhDAYDsfPzagl0u5wOhxuCoCJYUBT5TmchkeuIAl2t1suVaj4HlMtlFMVgGAKy%2bUKhXKpUS8USzfBQq1kqV4tgsVSuoCgm8EJL2%2bhsoihG4p1SuZxO5SiKIojuXb0E5cYATOKJ679bDYrCKQp/9WesQZQlRtU0gqpqrSJzskj3eV5VOEXmBrcrEqtIrCzSdwJKlliBgXWyyPBUk6NabK/J9ppcr8lTLZ5qsd0G1xe5XounWxzVv9rvxlEtnoZuNDylXR0oeaol0NDdgKeastDTUVgSzm1ipQOieohXDjplM1o0oUUTUTnAyma8coAWTVhJY/DyAVYyDfq0i/tYyazdUtY6DAPmNrhLtgJaSgmcTc28erg0851%2b%2bZHV8My0%2beRQ//TtxIM341%2b/n/3eY37l3HuxMvvNyvy3s68fvJv8xrjy5GjryeLMdyvz3%2b9vPG1l9e38NnLXMMDZTS2lpNGLcmzeZR493n52ZPg55JgK2if9lnGL4bl9d8S6M3JmnzpzTNl2njv3X9p2Rhy7L3zHkwHrhPtg1Loz4jK/gnJ6ZIgGyAIl0C1FQFURU0VU4dsyh8g8ooqYIqKqiMp8W2IRVcJUAdWgaRCZQ1ShrfUX2twd%2br3w4RiQ%2bK6OJUt4zcmiIQYJUnCgB51SsAYaCbDtIA2fki0/2w52W36qr2EGQAI0ojE0EqDhANHw3Tn8eN1DY2kdjcYLkamdlUf23RHf8VjUOeE7GgtYxnY3f9pff%2bS3vM6dvTnWP7n0zwctY0fbz4yrP1oMT9zmlx7ziHntkcf8MuKahvN6OL91t9BD2XW87tbR2AVwNrk8%2b83CuDaO7bvP99cf24zPtpb/9nbyoX75b77DFwtjDxymkbXZ75anv1uZ%2b3Zp%2bi9zo1%2buvfl2fe675ZmvTRs/wbktOLd5t9iCMqt43aNjiWIPDksUKJAZBkvQWJLpJJlOisPTPJFm8RSNJXkyzWBJFk/xhKbkiTSHpzniCmwnSaGJISGpZWSiwLRRnGb76cu/DCk34TTRxeFdw5b56HBp6X06mx9USVzFTFfMVfT8UVh6/UED/Q0zSJb/aMi3w2mq2WyRJAlBEMtzkixxHM9pm39a3CzwPM9zHMdrJXiS3Olg3V6XpuleVyNJkkVR1Nr%2bxpWqKlSvx/PCXYfTNpvrxOu1Wy1ulzMWiwcDp7ubGwcHVrvTeWjarzfgU5/n4MjSqNfPQ8Edgz54FkWRtt/rcdgddrsNzOcDJ66Nze1IOFoogDCC/PG5gfJLOC1LnCzLoiDSFMVx7CAX41iO4zhBEKX%2bpqog8DzH93rdwa8ratW5Wo7W7yPIsiQIPE0zWuVFP3P7Ix/9V8sqAkMAAFipVFCsg3c6rWarWq2gWJskSRRtE90ux3EMwxIESRBktVav12taVTBB3i4uunNSrgyQBYIisdX376zWo2OrFQCA7a0tu%2bP4wLS/vbXtdh6vrK6cBc8uLy73ds17hh3jzq75wGTa2zNu7zE0M7wEX/nAhZA2ihO4qBVVczzPBeMxX/winEnnIThWAH0XlxcFwBuNhbPZeLGYLBUFbbwOl5Rf3gDbw7wnfs03quVMNsfQrMXrXdzWL6yvHATCBqf9/a7x7fbW1PLaqsn0ZttgOT3VclLN1eV7ui6kXDMczw/V0f%2b%2baZTtYXar5TQYSueAVqNWKFTgFhQ/D2fzIIai2XS2AIKVchHIZ3PZbL1WJ/DOeSyWTCTTmczwx4DM4wxFWo8OQpHwkcXRatZj8WQBAAOBE5fHXwRLx0c2h8PudFjcHq/HbTUYd7K5jNvtAUAwHA7fAwMEgu5h6XS%2bUSvVqtVcKp3L5QulEgzBGNpOppI8x9VqDZphms2GwHOpxDmGU/dgdVG5NYgZIh6/uIhHz6OxaCCQSaUvE0mf11Uqlc6j5ziG4jiOoGixVCBwHMgmkTbxq4r/4RnAM6jAa9P5RzRYGdWKzSRpUAE7OJEwCPWk4ZMoySpHtXRMFyIJjKaZXo%2biaIqiqF6vR5Jkt6sd/CBJsndNA023q2mo4VOPohiKqPXzAZ7udHCGoQmCpCiKZdnf7TXf8rNf1jX7mv5KtvK7zEJaNLpt2HFYDvd29ncMW6dn2txyb3dmPhHMsTRRLJY6Hbzb7WJou91ucyzX62r%2bQ%2bB478qXiC5J0jQLNZuNZqODoiRBdLXzRRQEt%2bqVMgyjBI5jGNb3NrJRb3AcmwZygUQqXa0UECTfaiXL5VSlcp7NXoBgqlzKVSssx//zb4BkaWx/b8dudwcDfp/HfbC3XyyWTk/9NqvNYbXabPZyqVwAc9ZjUyR6GY9eeN1Oj9PVQdFkPBI%2bT9Sb9VT0dG/nwOc5iUcjB8eWbCbnstsBAHAHfK/frc5sbByHQpvW45mNzXd7u7Prm2%2bNhgX9ut5iJXuf2o74BwyAtXohWbxx%2blsHofqfKw12Jq%2b/QOBZ/uO69Y%2b/e1BNPuBFVREUSVBkbeAzjLauLUm8LAlahYamV/%2b5lJKnmjpJpAQW5RmMZzCB7dxmBLbTZzr8FYOJHC7x%2bC99bnW7gcQTWo4x4DlcA4srAikLhMT2xVsQ/yl0RJ7U8VSrB0d4IsV2Lhnsgulc9hmt5XBNww40WptgO5dUO852Ehz%2bSze2c9lD4jfoIrFbYmyAvvKTiP%2b/0YWjLFnWqhbL5zO%2b47H4yWwq%2bObSN3NxMpcJzMU80xHXVOp07uJkOuaZuvTNxDxT556pyuX7kH0sZJ%2b49M2mg28uTqajnulmeh0ZwsLWmlatQmMX%2beDExM%2bfjz77bOb1f2%2b9/X51/nvTxqPpF1%2bOP/tiZe7budEvxp5//mbsq1eP/2vyxeeH20%2bnfv7zy8efzY5%2bNff64eyrLydGvkyfvcUKBiinrfjdFbah7EZ/Za5fsYVWPFqVVsGBll3tshMpOeGiA6u6%2b4yzXXK2Nb2r3df3eY2BSxqPaDVcHqLuvVuc4DUXjSV/n8OgQwzqdJIkkSTJcdwgUOuvUkk3O9lC/6yDdHOG9Doy0Erx%2b0dNZXlw9GloFui63S5YKCAwdOrzeuy2Y5u7Ua3EImcn/vBFPOK0WvNAIZWIJVLZdCYH5kGvy2632YtgMXDiC/tdPn/A5/MVSpVhZQg6SZIQGGZZlsDxDtpuox2OZXu9bruNYiiKdzpa6Nnr4jhBEARNMwTeQVGU4zgtLtXuQAcxyNAMUPt0%2b7uHnWr9Y6QTBKFYAAuFIgzDjUaj1Wo1Go2Cdmy1AEFQvV6j6atw5aNhcLvQY4iRqw7HiUjgRG/Yc3t9Pq/XsLlpNh8emg/Mxp2tjc3z%2bAUMw/f5tegEQWjUa9rB4WKpVq0AAFgqlqrVaqVcKoBguVz9HfObP4L%2bD3UygD/xVNkKAAAAAElFTkSuQmCC' /%3e%3c/svg%3e)
- General workflow tuning
- HideDataset Action will no longer show in the workflow editor. The ability to manually create one of these actions has been deprecated in favor of the workflow outputs approach.
- Workflow run results can now be sent to a new history instead of the current one.
- Ordering of workflow steps is now sorted based on the layout in the workflow editor, arranged based on distance from top left corner of the editor. This won't affect existing workflows until re-saved.
- Workflows that contain steps expecting tools that are unavailable (as might be the case for a workflow imported from another Galaxy instance) will now have problem nodes marked with an error state. The workflow cannot be saved until the steps are removed or the tools are added to the current Galaxy instance.
Deferred Jobs & Managed Transfers
These components are under rapid development and interfaces should be considered experimental. They can be enabled by setting 'enable_beta_job_managers = True' in universe_wsgi.ini.
- Deferred Jobs
- A generic method for creating a dependency on an event before executing arbitrary code has now been defined in: galaxy-dist/lib/galaxy/job/deferred/init.py
- The deferred job runner loads plugins found in the same directory which implement the necessary methods check_job() and run_job(). check_job() returns a state which informs the deferred job runner whether it is okay to execute the run_job() method.
- The deferred job runner is independent from the regular tool-related job runner and is not coupled with tools, nor does it have any integrated cluster support.
- No documentation is provided for the format of a plugin at this time, but a sample plugin will be included at a later date.
- Transfer Manager
- Galaxy can now spawn persistent transfers of (unauthenticated) http and https URLs via the code in: galaxy-dist/lib/galaxy/job/transfer_manager.py
- The transfer manager is accessible in-application at app.transfer_manager.
- Transfers are daemonized and thus not influenced by Galaxy restarts, although a loss of database connection (restarting the database server) will cause transfers to fail.
- A transfer can be polled for progress via JSON-RPC requests to its socket. An interface for this request is available in the transfer_manager class.
- Future enhancements will allow for authenticated http/https and scp.
Updated & Improved
Data Content
- New "Get Data" source: modENCODE modMine server.
- New Genomes:
format: Genome [dbkey]
- Pseudomonas aeruginos (str. PA7) [16720]
- Pseudomonas aeruginos (str. PAO1) [pseuAeru]
- Pseudomonas aeruginos (str. UCBPP-PA14) [386]
- Silkworm (Bombyx mori str. p50T) [Bombyx_mori_p50T_2.0]
- Rice pathogen: Xanthomonas oryzae (str. KACC10331) [12931]
- Rice pathogen: Xanthomonas oryzae (str. MAFF 311018) [16297]
- Rice pathogen: Xanthomonas oryzae (str. PXO99A) [Xanthomonas_oryzae_PXO99A]
- Little Brown Bat (Myotis lucifugus) [myoluc2]
Current Tools
- Optional 'min' and 'max' attributes for tool integer and float parameters.
- Add --max and --un options to Bowtie.
- Validators enforce min/max values and generated error messages for invalid values.
- Add Standard deviation operation to grouping tool.
- Enhance histogram tool to allow plotting as frequency/counts.
- Allow fastx_toolkit clipper wrapper to work on fastqsanger formatted files.
- SAM metadata detection and setting sped up dramatically upon import.
- Wig-to-bigWig converter tool default parameters now same as UCSC's.
-
Cufflinks tool suite updates:
- Update Cufflinks tool suite wrappers to support v0.9.3.
- Add support for bias correction to Cufflinks and Cuffdiff; bias correction improves transcript quantitation results (FPKM values).
- Enable Cuffcompare to use sequence data so that it can generate data for use by Cuffdiff. Cuffcompare uses sam_fa_indices.loc to find locally cached genome sequence data and indices that are needed for this option.
- Add normalization support to Cufflinks and Cuffdiff and replicate support for cuffdiff.
New Tools
-
BED-to-bigBed converter tool now under "Convert Formats".
- Converts sorted BED files into UCSC's bigBed format.
- Requires bedToBigBed in PATH.
-
BLAST+ tools are now commented in tool_conf.xml.sample.
- Not in Galaxy main, for local instance use.
- If you run the BLAST+ tools at your site, please be sure to uncomment them if updating to the latest tool_conf.xml.sample (will be default at next update).
- Add SAMtools flagstat tool.
- Add CCAT ChIP-seq peak/region caller.
- Add BWA wrapper for SOLiD.
Histories
- Estimated size is now displayed for very large text-based (non-binary) datasets.
- Always show Galaxy masthead and enable Saved Histories to work with and without panels.
-
New "Copy Datasets" link added under History Options dropdown.
- Dataset and history IDs are now encoded.
- JS dropdown used to change source history, so you no longer have to switch desired source history to be "active".
- Remove link in "edit attributes" of datasets.
- Simplified destination interface by using a single select box by default, but also providing a link to show checkboxes for multiple destination histories.
- Use newly imported Inflector library to get correct plural/singular nouns on actions.
- Added arrow between source and destination.
- Removed checkbox for copying to a new history. Instead, create new history if new history field is not blank.
Data Libraries
- Creating job information (stdout/stderr) is now available on the library item info page, which is helpful when library uploads fail.
Trackster
- Add bigWig display to trackster. Automatically converts wig to bigwig if needed (NOTE: datatypes_conf.xml.sample has been edited to add the new converter, you must update datatypes_conf.xml to use it). The converter requires that wigToBigWig be in the PATH, but no other tools are needed to view bigwig files as they are provided by bx_python.
-
Tuning
- Fix track preferences not being applied
- Fix chroms not being selectable when a new track browser is created
- Fix ReferenceTrack not working with filters
- Fix visual analytics error when tool configuration has changed
- Fix visualization saving on Chrome by using $.each instead of for loop
- Fix shared visualizations
- Fix tracks fetching from data provider when indexer returned None, and when
- Fix BAM reads without cigar string.
Sample Tracking
- Additions and tuning to improve tracking (complete documentation will be available soon).
User Interface (UI)
- Version info is now printed in history item for Bowtie, BWA, Lastz, TopHat, Samtools, Cuffdif, Cufflinks, Cuffcompare, BFAST, and PerM.
- Turn off web browser auto-complete for tool search (includes workflows).
-
Grid changes resulting in better readability:
- Better page number display.
- Use "~" instead of "about", so "about 2 hours ago" -> "~ 2 hours ago".
- Cell padding decreased.
- Added new "nowrap" parameter to prevent text from being wrapped. Currently only used for tags to prevent "X tags" from wrapping in the middle.
Application Programming Interface (API)
- Still 'alpha', but: Initial pass completed for forms, request_type, users and roles. See README and examples in source.
Source
- Galaxy now runs with system python on 64-bit mac kernel.
- Enhance select parameter wrapper objects to provide access to additional fields by name for dynamically generated select lists (i.e. dynamic_options). \ example: use ${param.fields.path} to access a path field
- Updated the XML in filter specification for output files. The closing filter tag can now be on separate line to use as an # actual filter.
- Implemented 'from_work_dir' attribute for tool outputs. Using this attribute matches a file in the working directory to a tool output/HDA; when a tool finishes, the file in the working directory is automatically copied to the HDA. Hence, it's no longer necessary for tool wrapper scripts to manually copy tool output files to HDA files.
- More programmatic control of page numbers on grid (custom UI).
- Many formerly undocumented options have been added to the universe_wsgi.ini.sample file. Please compare your working copy with the new .sample and determine whether any of the options are relevant to your environment.
- The "welcome page" found at static/welcome.html, has been renamed to static/welcome.html.sample.
- The UCSC Genome Browser now supports loading data via https. If you implemented this former Galaxy URLs from https->http method in Apache as per the wiki documentation you will also need to remove it there, and allow the browser through your authentication scheme over https.
- Explicitly convert autocomplete dropdown values on refresh_on_change.
- Adjust image links in tools to work with a proxy prefix (thanks Brad Chapman).
Bug Fixes
- Ensembl GTF files are recognized correctly.
- Make Tophat wrapper compatible with python 2.4 by removing try-catch-finally.
- Fix unicode error for dataset peeks.
- Sanitize tool links in tool menu so that they can be searched. Use only lowercase letters, numbers, and underscores in links to ensure cross-browser functionality.
- Fix for building form element for boolean tool parameter when default state is configured as checked in tool configuration, but user provided value is non-checked. Fixes an issue seen in workflows that prevents saving the unchecked state under described conditions.
- Fix typo in BFAST.
- Trackster: fix BAM display bugs, Dense display bug, better dataset selection.
- Add validator to Tophat's segment mismatches parameter.
- Turn off web browser autocomplete for tool search in workflows.
- Fix Trackster's BAM display bugs, Dense display bug, better dataset selection.
- Add validator to Tophat's segment mismatches parameter.
- Added the ctypes egg so the DRMAA egg will now work under Python 2.4.
- Bug fixes to the Mac OS X launcher in the galaxy-dist/contrib/ folder (from Florent Angly).
- Python reports the wrong platform when running a 32-bit Python on 64-bit Linux, so Galaxy now forces the correct platform in this environment (thanks David Hoover).
- Fix Chrome not auto-saving changes to workflow checkboxes.
About Galaxy
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State.