-
Galaxy tools for the analysis of multiple alignments
- About the Alignments
- Whole genome alignments, even those produced by "quasi-global" algorithms, are inevitably fragmented owing to the complex evolutionary history of genomic DNA and karyotypes, which involves duplications, deletions, insertions and other rearrangements. These alignments also tend to be extremely large. To allow biologists to easily and efficiently manipulate multiple species whole genome alignments, they are stored locally at the Galaxy site, in a compressed form, and indexed. The majority of alignments are produced using Penn State's multiZ (for more information on multiZ click here) aligner run on the computational facilities of The UC Santa Cruz Genome Biology Group led by Jim Kent. Tools for handling multiple alignments described here are written by Dan Blankenberg and based on the bx-python package developed by James Taylor.
- Tools and Categories
- [Filters and Utilities](/main/MAF Analysis/#filters-and-utilities)
- For a list of data available through Galaxy, see [Available Data](/main/data-libraries/Available Data/).
- Format Converters
' width='665' height='589' xlink:href='data:image/png%3bbase64%2ciVBORw0KGgoAAAANSUhEUgAAAEAAAAA5CAIAAAAJJrqAAAAACXBIWXMAAAsTAAALEwEAmpwYAAAbUUlEQVRogZ16B3cbR5auftdLu%2be9mZ09u/bYlmXZVqDoseVdi0kaSZTEJCYQJAjmDBAkwAhmUIwScxBJiQQYxYgMdEAHNBrdABr5ncaVYVrWeHa2Th%2bcYrEa6Lq37r3f91VfsVgsXq/X6XTa7Xae5y0WC4qiXq/XYrGwLEvTtN1uDwaDBEGgKCoIgt1u93g8HMeZzWafz0fTtM1mEwTBZrNRFOX3%2b%2bFGhmHsdnsg4Hc4HAiC%2bHw%2bm83mchEMw9hsNp7jCIKw2%2b0%2bnx9BEIqifD7eZDJ5vRzDMCaTyefzIYnm9/ttNpvb7eY4zmq18jzvcrkQBBEEAUVRHMevOByOWCwWCAQ4jovH4wzD%2bP3%2beDyOYRjHcT6fz%2bFweDwem81mMpk8Hs/5%2bTmO4xRFIQgSCoW8Xi%2bKorFYDJYdiUScTmcgEOB5HkXRUCjsYVkPy8bjcZIk/X5/NBJ1OBzhUIj1sE6nMx6PORx2m90eDoVRFA0Gg5FI1OVywXzOKz4SgiA8zweDQfhFhmEIgoAJNE1fQVFUr9fLZLK2traenp6mRFOr1dPT08vLy6FQSCKR3L9///Hjx49%2bbtnZ2enp6UNDQ7Bgp9MZDoedTifDMMFgUDQwz7Msi6DoxMT4%2bvra3p7h/MJ4fna6trZ2eHB4cLBXmP80v6Cgp3%2bgpVWp7lDOzc%2b/e3esbFcvzy6eHB/v6HfevXvX3T1gMOhdJIWiqMfj8fv9CZcGKIrCMCwcDuM4ThDEFQRBlpeX29ratra2DAbD/Pz869ev3759u7u7e3R05Pf7m5qaMjMzHzx4kJWV9fDhQ%2bjfv39/cXHBZrM5HE6nwxEIBD7YQm6aRlBc2Vonl5VJykoKCwur5WX/mfG4q6v7zfa2UtGqUbXm5BdJJGXPcnJMJuPO9s4Pd9OluWWNdTVPnj3r6ur66%2bO80qLnI2PjNE1TFHV5CzmdTkEQEAQRtxBBEGdnZ83NzR0dHTqdTqVS9fT0jI6Ojo%2bPT05OhsNhqVT6008/ZWZmZmRkwGdWVtaPP/44PT21uLRoNJndbnc8HsdxnOf5eDye2DmhQCDgchEURRAufGZqfO/w6Ojo8PTs/Pz83Gyx%2bPw%2b0oUfvTsJhyP63d1gMMh5va83NhnafXZ2drB/YLPbjUbz3q6BomjYe5FIBMOwaDTKsixFUfF4nKZphmGu2O32nZ2dzs7O9fX1/f39%2bfl5MP/S0tLKygrHce3t7Xl5efn5%2bXl5eYWFhTk5OQUFBXl5%2bcvLSwaD4fziwmq1er1ek8mE4zjLshcXF5T44ITJZPL7/U6n0%2bkQo8KJOGk3DdHv9Xpxl8vhENMGjmEQ5Q6Hg%2bd5n89nt9tDwSCOYxRNB4NBs9lMkqTH4zGbzSzLYhiWiH5xPoIgV2iafvfunUwma25u1mq1zc3NKpWqu7t7fHxcp9PFYrHCwsLr16%2bnpKTcSrSUlJSbN29%2b/fXXOp0uHo9DVMXjcbfbHQqFILai0Wg4HGYTscuyLOf1xuNxj8cjCEI0GoX5giCA67xeL%2bQPBEFisVgoFMIwDKLL4/HE43GCIARBiMfjENwcx8GNDMOwLCt6QK/X9/X1GQyG4%2bPjlZWV3d3dk5MTg8FwcXFB0/Tk5KRWq%2b3s7FSr1b29vS0tLcPDwyqVamJi4s2bN2tra%2bC0%2bfn5tbW1N2/ezM3NbWxsrK%2bvz8/Pv3nzZmlpaXl5%2be3btwsLC2traxsbG3Nzc3DjwsLCmzdvVldXCYLged5oNHo8HrfbbTKZOI4DA/M8/3c8AMv1%2b/1gBrfbDWmUJEmwX05OjkajmZiYGBwcXFhYqKqqmpubI0myoqLis88%2b%2b/LLL6/%2bd9u1a9c%2b%2beSTjIwMgiACAcHlcsVisUgkguM4GNibcB14IBaLwfiHHoDsDoWM4ziLxYJhGMuyJpOJpmkcx4eHh7e3t7e2tlZXV/f29qanpw8ODiBsZDKZVCotLS2VyWSlpaVSqRQ65YkG4xKJpKysDDpSqbSiogLGpVIp/EuhUOr1O5ruXrPZ/IEHUBT9L3kAQpumaTA8rBvHcdjHMplMpVKNj49rtVqdTlddXT01NbWysiKRSL7//vu7d%2b/C5w8//HA30ZKd5DiMfP/998lOclpqaurz54U7WxsqVQdJ0eFwOBQKoSgKSQZiAMdxsQJGo/CoXq/3V1nIYrFcrs8Oh4MgCL/fb7VaWZYlSbK3t3dxcXF5eXl2dnZ9fX1wcFCv18/MzJSVlWVkZKSnp6elpSU/f9tJS7TL4/fu3UtLS4PPH3/8saio6Ojw4OTkBCogx3E2my0oZiGxTgWDQYACkJ38fj9BEL%2bqA3a7XRAEiqJcLlckEkFRFPKJw%2bHw%2b/00Tefn5zc1NY2MjHR1dQ0PD8tksrGxsTdv3lRVVeXm5j579iw7Ozs3Nzc7O/vp06fQycnJSY4/SbTkhJycnPz8/NLS0ufPnxcUFBQWFtbU1AiCEAwGYWPAVgkEAgiCYBgmCILVagUsZLFYOI6jKAri0%2bVykSQpYqGPLsDpdPp8PpZlrVZrPB7neR5Cx%2b12B4PBcDgMseV2u8W0HQrZ7Xa32y0IAvyMx%2bMBFIhhGI7jYBG3283zvFqtrqur6%2bnpAfBSXV3d19d3dHTU2NhYXFxcUiKW7ZKSkqJEKy0tLSwsTI6Xlpbm5ubu7%2b35fD4Mx8UF/P4WomkaXIFhmMPh8Pl8FouFpmmWZT9Ao3a7nabp36DRAIqiYEi73U5RFM/z7e3t09PTu7u7L1%2b%2b3N3dnZ2dXVtbI0lycHAwKysrMzMzLS0tMzMT9ltWVlZaWhqAgPT09MzMzPsPHpydnmj7tYvLqx4P8z6Ner1eMLCIARP51OVyhcPhQCAA0C8Z5S6XSxAEKOyQf6G%2buFwun88HMDYcDguCmBbBY8l65PP5wuGwRqPp6uoaHR0dHBwcHx9vbGycnJxcXFyUy%2bWAtTIzM2ElyU5WokEnLS3NZLwYGRpaXFoJh0NXoLCDgQF8A6qxWCwMw1AUZbPZLnvAarVCfjCbzRzHkSRptVphnCRJyHoMw9A0DeNOpzMJ6wmCYFm2sbFxenp6e3v75cuXkA9WVlYsFsvExERVVVVlZaVEIqmqqpJKpeXl5dXV1RKJRCaTyeVyiUQil1dWVVcf7u8ZDHqTyYzj2BWoDkkPgJfBYGBIiBiv1wsegMwQjUbhxqSLYONB1otEIsFgEG6EcvMLH/gNlOB5PhAIJJFCLBaDHwIOcBmkQH73%2b/3whe8LmdFoZFkWRVEwsM1mAypjNoswM2lgFEWBspnNZoqiPB5PgkB5CYKAKLJYLARBcBxnMpncbjdBEGazmed5hwi4xXxgtVoJgmAYsU55PAyGYeB8q9Wa4BIiVaJpEXuen58DhXI4HIAOcRynafri4sLtdjudTpvNlsQa7z3AcSKXu%2bwBSEqXPQAG%2b694IBwOhyMR%2bELa7YYOxEAoFPrgRtbrTQZPNBpNRleykME3R6PvmdqHUOIfjQHIQuCBZAxA8gFMBuM729sbW1uRqPg0BEkC1aTdbnAySbhOT0%2bNRiPPc/v7e7u7htPTs7OzcyidVouF9bJ4Is0Hg8LFxTnHcTz/Mw7HcXhUp9OJoqhIKSORiMfjgfoMcRaLxTAMCwaDPp8P1u3xeMAVOI4HAgEo%2bNFo1OfzgUVh2dFolKbp03dHQwPDvT3d2/o9g/7ty9lXh0fH%2b3uHJ0fvXs3Poyg2M6kbGtTKKyqWV19rNJ2jOt3szGxNVfXpueni7J22r3tHr9/dPzw9OX13dDg82Hdycka4CAhLWCRsFhFKwBYH0SEYDDqdTpIkA4GA3W6HbeNwOILBYLJQJFUJMDwUMqijUAcwDDvcM3Rp1F1qTV1d7fOSsry8wme5hZWV9TXlFaVlpbt7%2bxMTOm1Pd1FhYZtSVVNd82phcWxkKD8nt7d/YGd7669Z91pbm0vKKh/cf9Chas998iAr64Fa0%2bt2uyHqEAQJBoMoirpcriuwQ0iShHoJIgdUNAgMp9MZCoVIkoQVQmDxPG%2bz2QKBAMMwDocjWWih8gcCgcODfcPuHk3Tx8cnS4uLq%2btbxovzs9NTq9X27viE8TA2qxVBUBRDNze3nE7EZrMZjSbChbtcrq2tTYIkd3f3jo%2bP7Xb70eHB4dERgqCQDOBJgPeIpB4MTNM0QRCA%2bBiGCYfDkLxBNYGNAWENJEOkiImFieoDgkSjUQRBWJYV5QmHw%2bf3B0MhiFGGeV/IXK73hQxLfCHoLnFxczIMw8RiMYAkghAkCHGHeDweb0JWcblcYtpIRFEwGHS73cAcCIKgKErcQh%2bFEjabLQkloKwiCALqA1AN2HugfP1W2KJpOrFCUREjCAIM5vV6A4EAhmHJTRtP1AeGYaLRKChLbCI9QIxiGAaghqIor1dEqTzPwxb6RdhCECQcDjMMk4R4LMvC1wWDwSRSYFkvTYvJy%2bUiBEFMoyiKRCIRnuch60H1iEaj4BmGYQoKCmQy2d7eXmdnp1KpXFhYmJub6%2b/v39/fn5mZefr0qUaj0ev16%2bvrGo1maWlpYWEBEjeQY4qiIF3CgsNhUfkKh8Mejwd8CxNEMMclhD6wE3iA53kEQSCCbTYry7qdTpvFYmRZt9F45nKhFPW%2bflEUBZsePODz%2bYA3EQTxPNEaGxtra2sbGhpKSkpqa2uLiooUCsWrV6%2bKi4vlcnljY2NNTU1VVZVcLi8vL%2bd5HkAKSItAZQC8gM85jgNdKIm3ryT0vfjh4WFOTg6Yqqura2hoSC6Xv369vrHxtl1ZkJ/7P0qK/1hc9P/gKin%2bQ17O/5qa0iYKPnc5jV7WhcB%2b4NJ4PH5xcZFkWFAZIXEDB4zFYggiuvSjjCwQCCQZ2Ye6kNFoFATBYDC0trYeHBysJ9rZ2dnLly8vLi5OT8%2bVyqp7P/1bRsbn6emfZWZ8np7254yMzzMyPp%2bdeWG1iQj598FcUqN1Op2QucF1yQrodDpxHAfwAjgcMAjEAJTORAyIevNHChmCIPF4/OTkpLS0tDfR%2bvv7x8bGampqNjZej%2bkmFW35TY3XWlputTTfaG252dx0o631Vl3ttZEh9dT0rJCgLH9DmXMl7QQlkuf5pOwDmSOemACl83c88LeUOY/HI0KJYDB4cHCgUCj0ev3q6ury8vLBwQGoDxMTk0ND3cXFGUVFmYXP0yWlDwoK0kpKsqTSh8tLc6urK7SbtlrMPh9vtVpIkkgUfBPDuGmaslotPh/vdDoQxOn3%2b%2bx2K0G4PB7GYjHzPEcQLqvV4vf7HA47cAnA5yzLWiwWv98PSQai6wN5/UNOHI/HDQZDRkZGQ0NDa2trc3Nzd3d3Xl7e0tJif/9wbe39mpp/baj/vK720/q6PzfUf1Zf9%2bcyyf99Md6bALdBggADuwMBEfTiuKjMhUJhkhTtxzBelhXTOUWxgYCozLkS8wMBgSTFIPF4vAzDQP4IBoNQYaLRKKjnINz/nrxusVgEQTAajUtLS8CAATafnJxQFEXT9KtXc11dvSqVWqVS9/Zq6%2bqatNqhnh7t%2bPjg7EzjxETt2Gjl7Ezj2GjlxETNzHTD6IhsarJuMjE%2bM9OgG5OP66rECWPyiRc1U5N14vh0w8SLmrFR%2bcx03dRkJ0nSQKZ5nodsA%2bUC2J/dbmcYBmo/FJBfQQkoQ0nageO4x%2bOBpAtY%2buHDhwqFYnR0VKPRzMxMS6XS2dkZhwNVtMlznv3pecGn%2bXmfFOR/kp/37wX5H%2bnk54mX2EkMXu4U5H%2bal/un0pLvCRF3ko6ETA/ySTgcdrlcFEUBjIUKCCUV5LZIJPJelQAoljw1QFGUTmjCAJ7dbvfk5NTR0ZHBYNDr9ScnJ4uLi%2bfnZ9vbOz09iuqq7ytldyrKb1fJv6sov10pu1Ml/65celtemfrLeEWKrCIlOUFemfrLhIqUivKbSmURQVBDw8Orr7diCYiRXAD5Mw7/Owv4fV2opKSkvr5%2bYGBAqVT29fVJJJLR0dGFhaXWtiqJ5HZZaUpJ8S1pWWpJ8S1JaUqZROyUSe5IEuNlktTSktulJbfFCSUw4U5JyfsJiX/drKp6RJNkXU315tudSCQMwhbwHpIkw%2bEwwMfLJzQ4jgO/ea8LfRTMOZ1Ov9/PMMz4%2bPjm5ubW1tbm5ub%2b/v7U1NTBwf7a2oZKVSeXf11Rca28/JpM9lV5%2bZcVFYmO9EtZxVeyikRH9lVF%2bbWKimuVldffT6j4Siq9WlEu3lUuvSYt%2b6y5ORvHXIeHhyaTKRqNJPNsUs7AcVcgEADRF9Jo0gPvwRzodRiGXYbTEMokSebl5TU0NAwMDCgUit7e3rKyMp1ubH19o62ttqjwu4KC1Ly8lMLCv%2bTl3i7Iv1NY%2bJfc3NvPn/8ynp%2bXkp8Yz8tLKShIff78u%2bLiH2QVaaUld0tKfiwt/aG5qcjvF3Uah8NBkjSCYO/eHZMkfXFhMpstFOU%2bPj51OBAcJ46PT3GcQBAU4Korgb1FKBEKhYC/g4Tt8XgANsPRpcPhiMfjIPqBQJRgvBHIZeFwxO0Wo5%2bmPaAdEASdOOCIMIxY11hW5IOQLgUhGIvFxsYmtdqRqam5wUHdzMxCS4tiaGhof/%2bovq6srvazpsYvGuq/aGz45RL/TIw01H/R0vSFVPrZ9tvN05NThxNh3O73HiBJ8rceAEKDomggEMBxHDaV1WplGCahVIr1i6Ypm80qCAGbzUpRpN/vs1jMLOthGLfdbg0E/CjqxDBUEAJ2u42iSJ7nlErF%2bLhOr9%2bZnZ3W63dmZmZWV1coin7xYvTpk7/89cGNrMxvHj28fT/r2wf3bzx6eDsr65u/Prj58OGtROfGs5z/sFktne3tS6uvvV5WjAHwAEEQlz0Az81xHBwDA40AmO3z%2bQRBgNPV3z8nhnpEURSQbK/XKwiCRqPp6ekZHh7WarVjY2MNDQ2TkxOvXi1UVhY2Nlxtaf6yqelqc9OXyevyn22tX1aUf7H95rVudGT/4Ijzev%2bbnJjn%2bY9y4kAgAGd%2bHo8Hsh4gAsjLNC0e8ikUipcvXxoMhrm5uYODg1evXq2trVks1qWlxcHBrp6edqWyQatVd3S0dHa2Dgyo29sbu7uVfX0dSmVjb69Kq9UYLy4YhsET7R%2bglBD7SUoJrktOSFJKyL/gOggVOPaDg59AIADCG%2bToeAJX8zwPLgJljqLEcY7jAwHhsjLHMGKw%2bXw%2bAHPvs9DfUqeTlBIKRZJGJCmlxWL54F0JWLnJZAJBCUxz%2bV2JhDInvgrh9XpdLhc43263w%2b4CFO12u43GC6/Xa7fbEQThOM5oNILYkzxi%2bhWlhK18mVJ6PB7Y64IgiOe4Px%2b5QZCgKOr3%2bwFaRSIRjuPAcq5EAyaQAHMhEAChAUkHSQ%2bIDtTUeMIVcIoKJwzhcBgmsCwLYiggA4DZlyklSZIipfy7MQBaQJJKf1QXAnCrUCgKCgrq6%2bsVCsXU1FR/f//Tp0%2bViabRaPr7%2b9vb2zs7OxsbG3U6nVKp1Ol0GxsbNTU1ra2tWq12fn7%2b0aNHKpWqpaWlq6urt7e3sbFRo9HU1dVNT0%2bD8y8/6nsw97fqQDILYRiWzEIfjQHgGQRBrKysjI6OwmGUTqfb3t6emJhobm7u7OyUy%2bXAV/v7%2bwcGBiYnJzUazdTU1NjYWH19fUtLy%2bLi4snJCSjsbW1t3d3dAwMDo6Oj/f39zc3NExMTkN8%2bAqeTwhbUAafTeTkG3G43HLkRBIHj4oNiGOrz8QmTOyORCMt6HA57OBxyOh08zyXkGjHbBoMChonECjJSKBSkabG8RCIRkiRiMVGTpCjS5%2bNZlhGEQCwWs9mswWAwIQjAyQjNsmLpJElSEAKJV4/EF5AoikRRJBQKfyhsQZpPiswEQYDMn1StzWar04nt7R2azTa73WkwHNjtKILgKOriuIDJZMNwimX9Z2cmkmQIl9tksnm9fovFYbejfn/IZLKhKEnR7Pm5hWV9KEoajVa/P2SxOB0OjGV9GEZwXIBhuIsLC8cFxC934jwvmEw2gmBY1mcy2VjWR5IMSVLwqGIWAoUQPBCPx3d2dqqqqtRqdXd3t1arValUUql0b29Pqx2UV95uV37d2vq1ou0bRds3ba1fq9q/qaz8ulx6Q6NObVfeVrXfUanuKJUpKpXYaU902ttT2tsTHWWKqj2lo/39uKo9pV2Z0pEc70gtKkxtqL/Z2vJtc9O3LS03mpu/bRZZuNhJ0PEbzU3fKhW3ZBVXdw1vQC8SPZCE05Bttra2mpqadnd3Nzc39Xr97u6uTqfDMGx3d1ciefTs2e3s7FvPnt3JyUnNzr719OltqfRhQX56etrVtHtX09OuZWZev/fTFxkZX2WkX0v76Wpm5vW0e1fT0r6ETnr6tYyMr3766YvMjOvp6dfu3RMn3BNvFCdUyvJqqrMryjMlpfcqZQ/KJOlSaYZc9kBSeq%2biPLNSdl8iSausvF8myTg%2bPgTu9Ss%2bAAvY29trbW3t6%2bsbHBwcGhpSq9UNDQ0Gg16hUErLbjU2fFVXe72%2b7npD3fW62q8aG65Ly74ul97UaL5LmDO1syNV1X6nQ5XaAZ2OVPAG/JnsdHSkdohOuJOc36680dJSotefvH69PTU9bzAcLS9vrK5u7e0dz82tbG0Ztrf3Z2eXDIajlZVNs9kSj8fe84FkIQMq/fbt2/b29rdv325ubm4n2vT09Onp6fj4i9raguLiu4XP7xYX/0dJyX8%2bf/5DUdHdmppcieRRdvatx49vPcm%2b/exp6qNHN58%2bufPkScrjxzefPUt9/PhW9uNbYufRzSfZt58%2bvSNOeJr65Mnt5IQnT1Kys2%2brVI2Li9NTU0NDQ5q5Vzqdru/FeP/c3PjISPf01NDL2dHh4a5XL0dfvBg0Go2AO99ro9FoNHl%2bsbu7W15e3tLS0t3drVarm5ubi4uLt7e3a2pqHz/%2b1%2bf5/7sg/5/z8/4pP%2b%2bfCvL/uSD//2Q//kNuzr/X1X5SJf%2bkukq8qhKf/%2bhVU/3pgwefSkr/IJf9sVL2L7KKP1YmLrEj%2b5dkp0r%2bp9yc/2kwbCRVYfFtleQRUygU2tnZGRwcNJlMe3uiPH9ycrKxsYFh2NnZ2eLiwuLi/Pj42KtXswsLc2NjIwsLc2trq2Njg4ODPWq1oq9PPTzc19HROjDQ3d%2bvUavbhof7urrau7tVIyP9MGFgoLuzs21oqLevT61WK0ZG%2bru6lD09HcPDfZOTOp1uSKvt7uxoGx7u7%2bpS9XR3jIxoOzsV/f1dg4O9nZ2KwcFerbbHaLwA1CMqcyCPgap6%2bbVLQASgx1%2bewLIi0AD1OEFoRBgCNwLkoigxxwHKSGI16ACU8HhETAZSyGUoAeXpd45ZWVZEFvACxM8HCN7/D0UmW2m2Dy4sAAAAAElFTkSuQmCC' /%3e%3c/svg%3e)
- Alignment Extractors
- Alignment Stitchers
- The FASTA output is relative to the genome of the input interval; positions that do not exist in this genome are not included. Overlapping blocks are split at the edges of their intersections, and the block fragment with the highest original score is used. Users can also select which species to include in their output.
- Filters and Utilities
Galaxy tools for the analysis of multiple alignments
For the latest information on the MAF tool suite please see: http://usegalaxy.org/u/dan/p/maf
If you use these tools in your research, please cite: Blankenberg D, Taylor J, Nekrutenko A, The Galaxy Team. "Making whole genome multiple alignments usable for biologists." Bioinformatics. 2011 Jul 19.
About the Alignments
Whole genome alignments, even those produced by "quasi-global" algorithms, are inevitably fragmented owing to the complex evolutionary history of genomic DNA and karyotypes, which involves duplications, deletions, insertions and other rearrangements. These alignments also tend to be extremely large. To allow biologists to easily and efficiently manipulate multiple species whole genome alignments, they are stored locally at the Galaxy site, in a compressed form, and indexed. The majority of alignments are produced using Penn State's multiZ (for more information on multiZ click here) aligner run on the computational facilities of The UC Santa Cruz Genome Biology Group led by Jim Kent. Tools for handling multiple alignments described here are written by Dan Blankenberg and based on the bx-python package developed by James Taylor.
Tools and Categories
[Format Converters](/main/MAF Analysis/#format-converters)
[Alignment Extractors](/main/MAF Analysis/#alignment-extractors)
[Alignment Stitchers](/main/MAF Analysis/#alignment-stitchers)
[Filters and Utilities](/main/MAF Analysis/#filters-and-utilities)
For a list of data available through Galaxy, see [Available Data](/main/data-libraries/Available Data/).
Format Converters
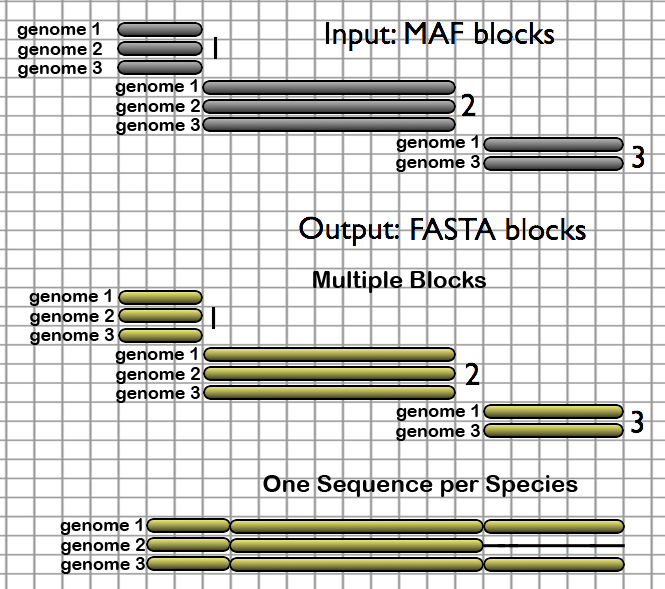
These tools convert MAF formatted files to FASTA and offer the ability to restrict alignments to a subset of species. This is useful for performing further analysis with practically all currently available genomic tools.
Available Output Types:
- Multiple Blocks - create one FASTA alignment block per provided MAF Block; blocks missing a desired species can be kept or discarded
- One Sequence per Species - create one sequence per species, where all MAF blocks are concatenated in the order and strand in which they appear in the selected source MAF; desired species missing from a particular block will have their sequence padded with gaps
Alignment Extractors
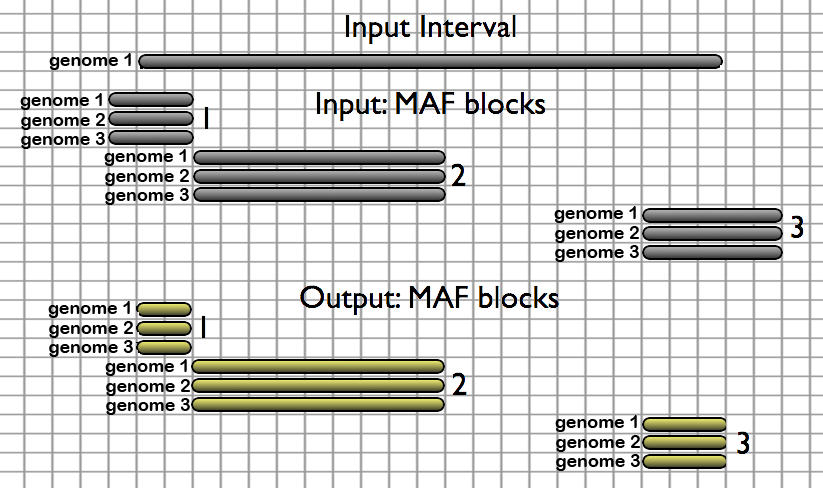
Extractors take genomic intervals as the input and return pairwise or multiple alignments corresponding to these intervals as illustrated below. Users can use locally stored ([cached](/main/data-libraries/Available Data/)) alignments, or alignments from their history as the MAF source. Here, three MAF blocks overlapping a single interval are extracted. MAF blocks are output relative to the strand of the provided interval, where the default is '+'. Blocks 1 and 3 are trimmed because they extend beyond the boundaries of the interval:
Currently there are two types of extractors that may be merged into a single tool in the future:
- Extract Pairwise MAF blocks given a set of genomic intervals - takes a series of genomic intervals and extracts pairwise alignments from a large selection of locally cached MAF files.
- Extract MAF blocks given a set of genomic intervals - takes a series of genomic intervals and extracts multiple alignments from a large selection of locally cached MAF files or from alignments in the user's history.
Alignment Stitchers
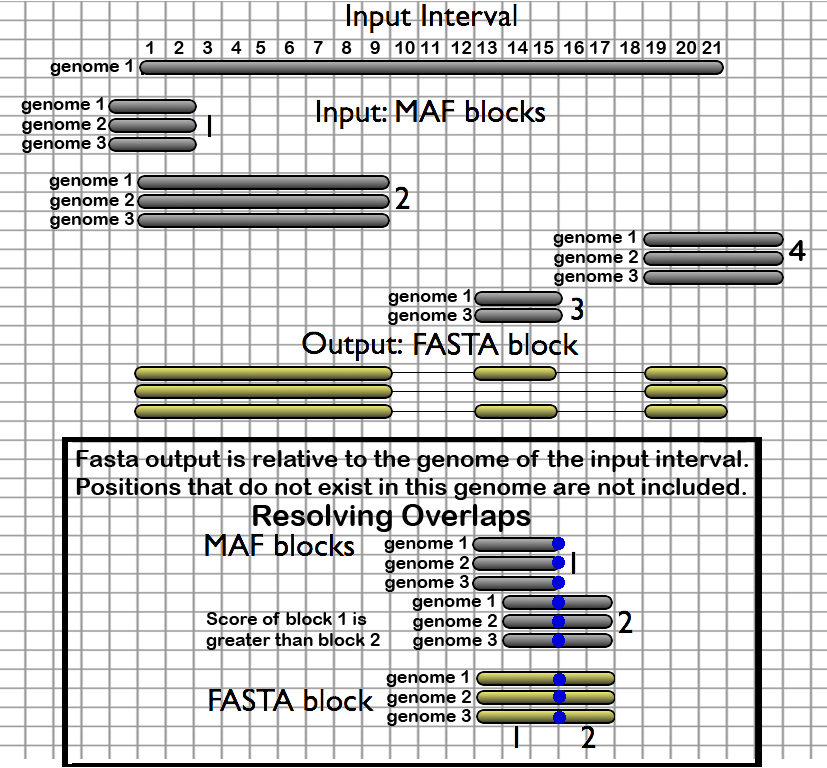
Multiple genome alignments consist of a very large number of relatively short blocks. This is why extractors (described in the previous category) typically return multiple MAF blocks per interval. In many cases, however, it is desirable to join (stitch) these multiple alignments together into a single continuous one. This is the purpose of this category of tools. The figure below shows a genomic interval overlapping four blocks. Stitching these together returns a single alignment (in FASTA format) where the void between blocks (2,3) and blocks (3,4) is filled with gaps:
Currently there are two tools that perform this operation each able to use cached or user-supplied alignments:
- Stitch MAF blocks given a set of genomic intervals - creates one FASTA alignment block for each supplied interval
- Stitch Gene blocks given a set of coding exon intervals - creates one FASTA alignment block for each supplied gene (requires a gene (12 column) BED file), where each exon interval is processed as above, and then concatenated together
The FASTA output is relative to the genome of the input interval; positions that do not exist in this genome are not included. Overlapping blocks are split at the edges of their intersections, and the block fragment with the highest original score is used. Users can also select which species to include in their output.
Filters and Utilities
-
MAF coverage statistics - computes the coverage of a genomic interval by a specified alignment; if a column does not exist in the reference genome, it is not included in the output.
Consider the interval: "chrX 1000 1100 myInterval" Let's suppose we want to do stats on three way alignments for H, M, and R. The result look like this:
chrX 1000 1100 myInterval H XXX YYY
chrX 1000 1100 myInterval M XXX YYY
chrX 1000 1100 myInterval R XXX YYY
where XXX and YYY are:
XXX = number of nucleotides YYY = number of gapsAlternatively, you can request only summary information for a set of intervals:
| #species | nucleotides | coverage |
| hg18 | 30639 | 0.2372 |
| rheMac2 | 7524 | 0.0582 |
| panTro2 | 30390 | 0.2353 |
where **coverage** is the number of nucleotides divided by the total length of the provided intervals.
- Filter MAF blocks by Species - restricts alignments to a subset of species
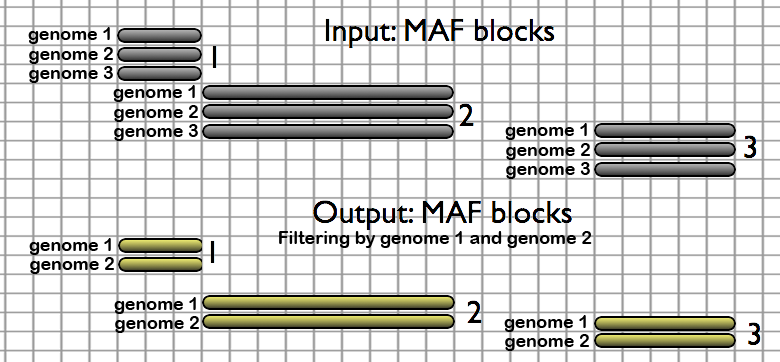
Users can choose to keep or discard (1) blocks which are missing a desired species and (2) blocks which contain only one species.
- Filter MAF blocks by Size - restricts alignments to a specified size range
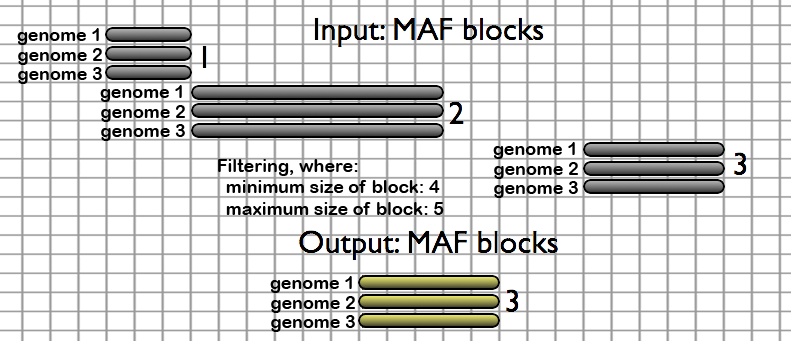
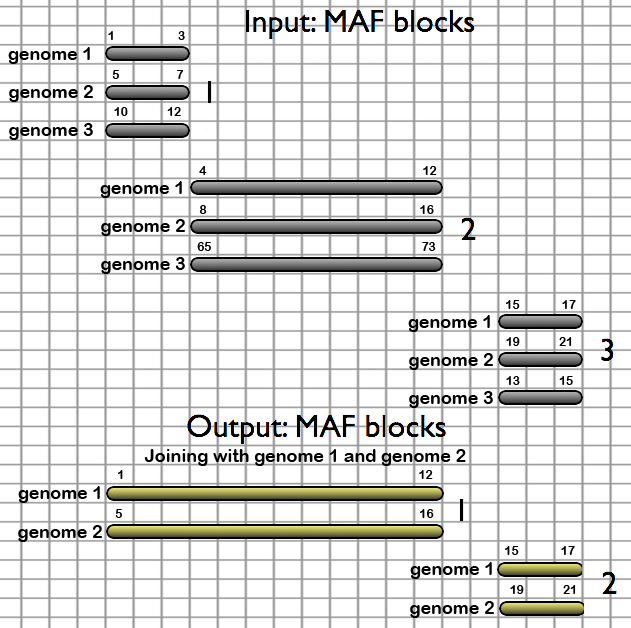
- Join MAF blocks by Species - Merges MAF blocks which are adjoining in each specified species from a MAF file. Columns which contain only gaps are removed. Species which are not desired are removed from the output.
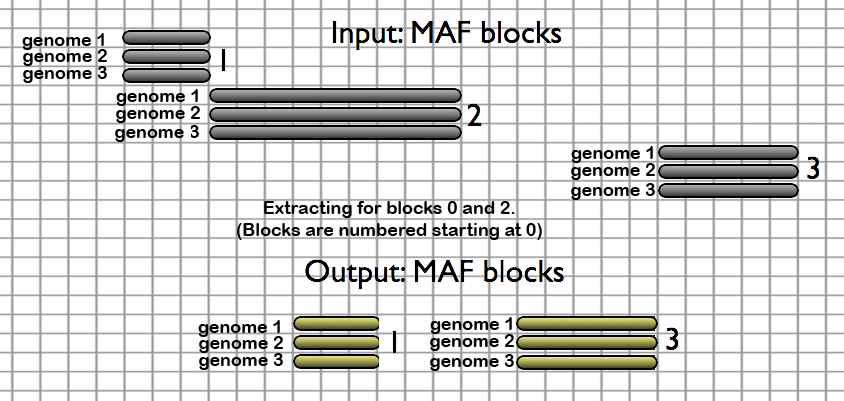
- Extract MAF by block number - extracts specific MAF blocks by their number in the dataset; block indexes start at 0
- Reverse complement a MAF file - computes the reverse complement for all blocks in an alignment
This MAF Block:
a score=8157.000000
s hg17.chr7 127471526 58 + 158628139 AATTTGTGGTTTATTCATTTTTCATTATTTTGTTTAAGGAGGTCTATAGTGGAAGAGG
s panTro1.chr6 129885407 58 + 161576975 AATTTGTGGTTTATTCGTTTTTCATTATTTTGTTTAAGGAGGTCTATAGTGGAAGAGG
s mm5.chr6 28904928 54 + 149721531 AA----CGTTTCATTGATTGCTCATCATTTAAAAAAAGAAATTCCTCAGTGGAAGAGG
becomes:
a score=8157.000000
s hg17.chr7 31156555 58 - 158628139 CCTCTTCCACTATAGACCTCCTTAAACAAAATAATGAAAAATGAATAAACCACAAATT
s panTro1.chr6 31691510 58 - 161576975 CCTCTTCCACTATAGACCTCCTTAAACAAAATAATGAAAAACGAATAAACCACAAATT
s mm5.chr6 120816549 54 - 149721531 CCTCTTCCACTGAGGAATTTCTTTTTTTAAATGATGAGCAATCAATGAAACG----TT